預測流感病毒的演化
我們經常聽到有人問:基因突變不是隨機的嗎?如果是隨機的,病毒演化可以預測嗎?事實上,病毒演化不同於基因突變。突變是指病毒在複製過程中出現的隨機性錯誤,它沒有特定的方向。而病毒演化則是由病毒突變、宿主免疫反應、以及環境共同構成的一個複雜過程。因此,雖然病毒基因的突變難以捉摸,我們卻可透過模型追蹤宿主中優勢突變的動態來預測病毒的短期演化。正如儘管我們不知道風從哪裡來,但我們可通過觀察受風影響的物體來捉摸風的方向和力量。同理,宿主的免疫反應和環境增大病毒基因改變的壓力,其中一些關鍵的突變會體現出人體免疫反應的選擇趨勢和病毒的傳播情況。因此,透過研究病毒與其宿主共同刻畫出的歷史數據,我們便能夠了解病毒的演化特徵。
鋪墊了病毒演化預測的基本原理,建立病毒演化的模型仍是一份艱巨的工作,這需要收集大量的數據將病毒基因組和全球各地病毒流行的狀況串聯起來進行分析。比如,要捕捉H3N2病毒(季節性流感的其中一種主要亞型)演化的模式,我們要收集20 年間的基因數據以及十多個國家疾控中心(CDC)發表的流感監測數據,並從中學習。對新冠病毒,我們需要研究分析上百萬個病毒基因組。在演化生物學中,根據物種基因組之間的相互距離對物種進行聚類而建立的系統發育樹(圖 1)是一種經典的分析工具,它能清晰的展示毒株之間的祖源關係以及病毒的演化歷程。通過對系統發育樹的分支優勢(Luksza 及 Lassig,2014)或分支進程(Neher 等人 2014、Huddleston 等人,2020)進行建模,研究人員提出了幾種方法來預測病毒未來演化的趨勢。然而,我們採取了另一個角度來進行有關病毒演化的預測,不是如系統發育樹般把整個基因序列當作基本分析的單位,而是把核苷酸位點上的演化動態視為模型要捕捉的主要目標。這種方法源於觀察,即關鍵突變往往分階段發生,部分有利的突變在疫情爆發高峰前幾年已經出現,其餘的則在隨後幾年和高峰季節期間發生(Wang、Lou 等人,2021)。在宿主個體內,數十億計的病毒被複製,產生有利、有害和中性的突變。在宿主免疫的高度壓力下,携帶有利突變的病毒能避開如抗體識別的免疫機制,並大量複製。那些携帶重要突變的病毒更有機會傳播給其他宿主,其後產生的突變(亦稱為「尾隨」或「輔助」突變)常常具有維持病毒的功能或穩定其蛋白質結構的功能,促進主流變種病毒的形成。因此,在主流毒株形成以先,病毒已經在殘基置換下開始進行競爭和選擇。這表示,建立基於核苷酸位點的演化模型可能會讓我們更早找到影響演化歷程的重要突變。我們提出了幾個新的定量來量化這個過程,其中包括突變的有效期,它描述新變異在人群免疫反應下保持優勢的時間;以及突變爆發的門閥值,它描述突變體的適應性到達何種程度時該病毒會引發新一波的大量人群感染。基於這些以及其他信息,我們開發了一套預測模型beth-1,找出能夠代表未來病毒群組的最佳毒株,以作為製作疫苗的抗原藍本(Lou 等人,2022)。該模型所預測的疫苗株(如按設計生產),預計可使現有疫苗對 H3N2 的保護效力升高絕對11.2%(95% CI:3.5-18.8)。我們亦正為 SARS-CoV-2 病毒的演化開發預測演算法,目前數據顯示,該模型能夠在 Omicron 變種病毒在人群中成為主流病毒的 6-12 個月前,找到關鍵突變。
預測疫苗的效力
在2021 年初,先生問我:「最近在忙些什麼?」答:「讓人非常振奮的工作!我們找到一種方法可以在人們接種疫苗或感染病毒以前,預測疫苗效力。」他道:「匪夷所思!」2022 年,我們研究小組發表了全球首個透過基因組分析來預測新冠疫苗效力的方法(VE-GD 模型),在獨立的數據組中預測準確度(一致相關係數)達到 95%(Cao 等人,2022)。如要從臨床試驗或觀察性研究中取得疫苗效力的黃金標準測量值,通常需要較長的時間來進行疫苗接種、人群感染、臨床診斷和數據分析。如果可以用電腦模型來估算疫苗效力,便可迅速評估疫苗在新變種病毒挑戰下的保護性,令政府官員盡早知悉新變種病毒對衛生系統的威脅,以及時制定適當的公共衛生政策。此外,疫苗生產厰亦可根據疫苗效力的預估結果,設計出更優化的抗原和臨床試驗。
這個VE-GD演算法是通過統計模型實現的,它將病毒突變所引起的分子水平變化以及疫苗在人群層面所產生的效力連繫起來。雖然聽起來很簡單,但要完成這個設想卻並不容易。首先,我們需要找出對疫苗保護力有影響的突變。在對流感疫苗的研究中,我們通過統計或機器學習中稱為特徵選擇的方法來解決這個問題。在新冠疫苗上,我們發現對疫苗效力影響最大的基因組區域是刺突蛋白上的受體結合區,它正是宿主免疫識別的主要目標。另一個研究的難處是疫苗數據的質量,由於各國對流感病毒基因組的測序數據零散稀少,疫苗有效性的研究欠缺標準,因此疫苗效力預測模型對流感病毒 A /H1N1pdm09 的最佳表現僅為55.0%(R方),對 H3N2為 87.8%(Cao 等人,2021、Cao 等人,2022b)。在2021 年,隨著大量關於新冠疫苗有效性的優質報告的出現,模型的準確性得到了大幅上升。然而,新的挑戰出現了。不同於流感疫苗采取相當單一的技術,新冠疫苗具備多種疫苗技術平台,例如 mRNA、病毒載體,以及蛋白亞單位疫苗,因此疫苗有效性出現了更多變化。於是我們引入了新的方法來解決這個挑戰,最終建立了一個相當精準的預測模型。比如,該模型預測 mRNA 疫苗針對 Delta 變種病毒有症狀感染的疫苗有效性為 82.8%,在實際數據中得到的疫苗效力為 83.0%(圖 2)。
這些預測方法構成了我們在生物科技領域許多努力中的一部分,即通過生物信息學以及統計學方法助力疫苗研發。精準的計算和預測疫苗抗原表現,能夠幫助我們從反向疫苗學的角度設計出更優越的疫苗抗原 —— 預測未來,以引導當下。
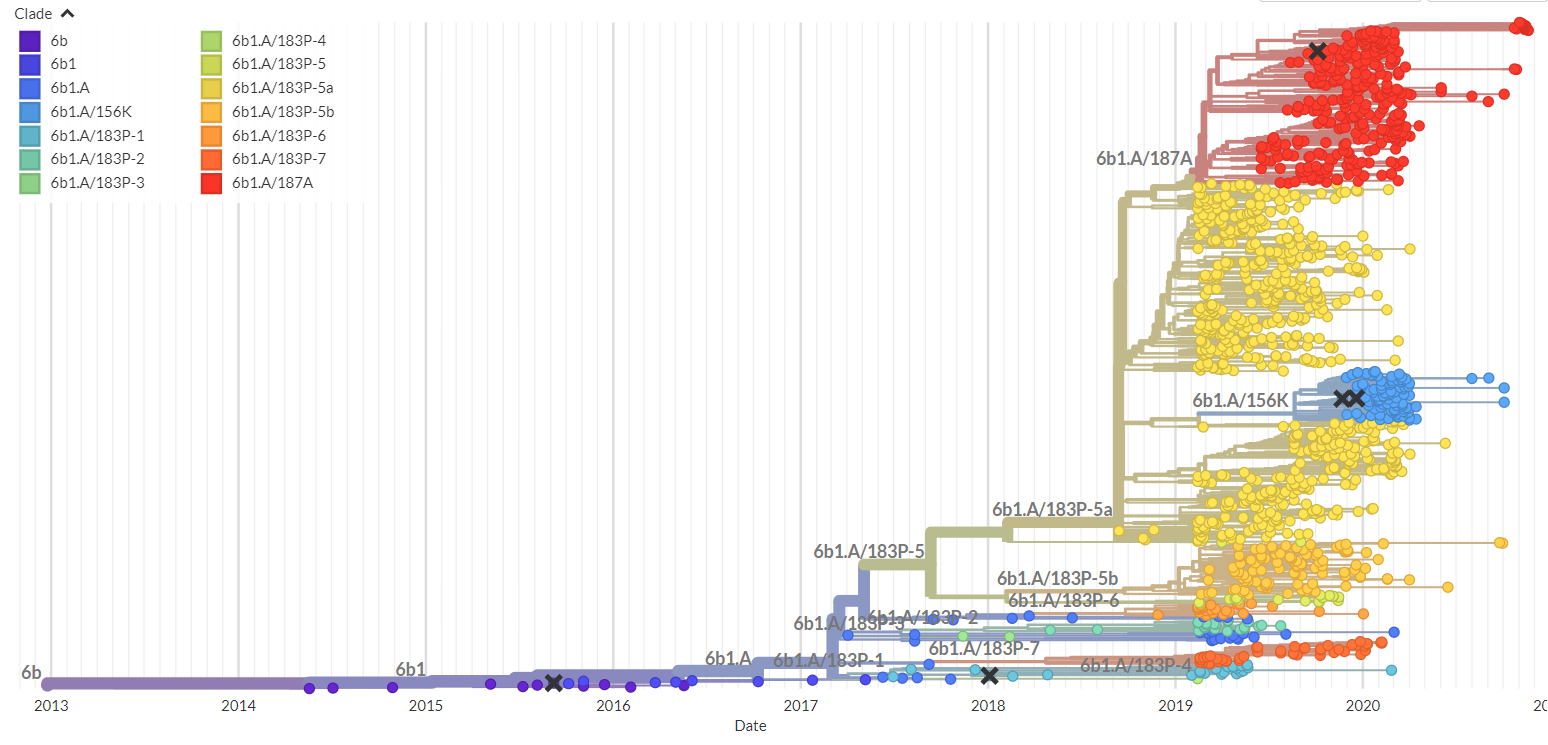
圖1: 由Nextstrain繪製的SARS-CoV-2病毒系統發育樹 (https://nextstrain.org/)
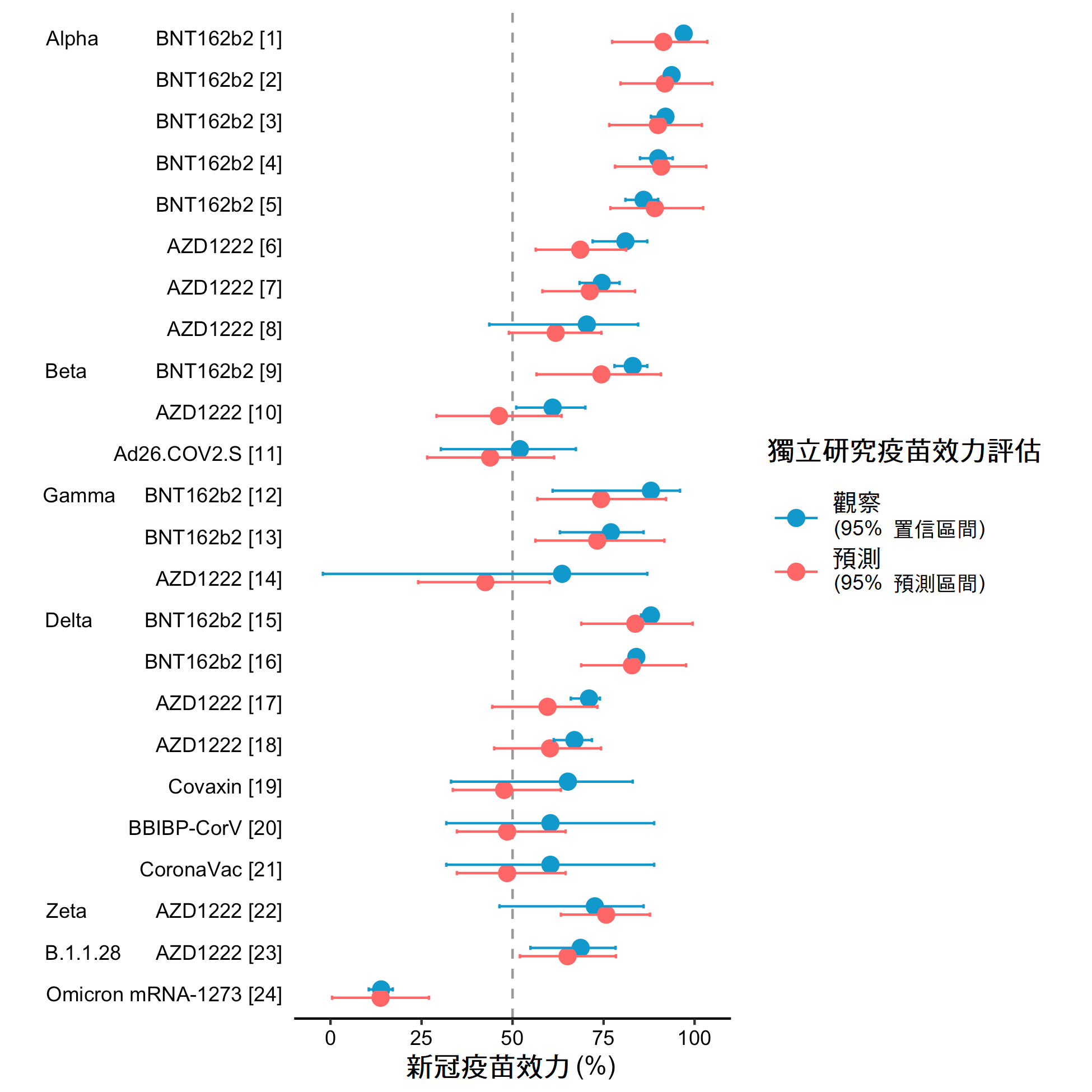
圖2: 預測及觀察新冠疫苗的疫苖效力 (來源︰Cao等人, 2022)
參考資料︰
- Cao, L. et al. Rapid evaluation of COVID-19 vaccine effectiveness against symptomatic infection with SARS-CoV-2 variants by analysis of genetic distance. Nat Med, doi:10.1038/s41591-022-01877-1 (2022).
- Cao, L. et al. In silico prediction of influenza vaccine effectiveness by sequence analysis. Vaccine 39, 1030-1034, doi:10.1016/j.vaccine.2021.01.006 (2021).
- Huddleston, J. et al. Integrating genotypes and phenotypes improves long-term forecasts of seasonal influenza A/H3N2 evolution. eLife 9, doi:10.7554/eLife.60067 (2020).
- Luksza, M. & Lassig, M. A predictive fitness model for influenza. Nature 507, 57-61, doi:10.1038/nature13087 (2014).
- Neher, R. A., Russell, C. A. & Shraiman, B. I. Predicting evolution from the shape of genealogical trees. eLife 3, doi:10.7554/eLife.03568 (2014).
- Wang, M. H. et al. Characterization of key amino acid substitutions and dynamics of the influenza virus H3N2 hemagglutinin. The Journal of infection 83, 671-677, doi:10.1016/j.jinf.2021.09.026 (2021).
作者︰
香港中文大學公共衛生及基層醫療學院王海天教授
2023年1月