Predicting Evolution of Influenza Virus
A common question we often encounter is: is mutation of virus truly predictable, and isn't it random? Well, the short answer is, virus evolution is not equal to mutation. Mutations are random errors occur during the replication process of virus, and they do not have a preference on their destination. Yet virus evolution is a complex process shaped jointly by virus mutation, the host immune response, and the environment. Thus, although mutation at given positions in virus genome is unpredictable, short-term virus evolution might be projected by tracking the dynamic pattern of advantageous mutants in host population. This is like even if we do not know where the wind comes from, we can tell the direction and strength of wind by observing objects influenced by it. Similarly, host immune response and the environment exert traceable pressure on the virus genome, and key mutants exhibit patterns of immune selection and virus transmission. Characteristics of virus evolution are thus carved in the history data written down together by the virus and their host counterpart.
Insomuch as we have laid out the rationale of prediction, building models for capturing the pattern of virus evolution could be strenuous, as a huge amount of data on virus genome and associated epidemics worldwide would need to be linked and analysed. For example, to learn the pattern of virus evolution of the H3N2, one of the major subtypes underlying seasonal influenza epidemics, we collected genome sequencing data available in the past 20 years and epidemics information in over ten geographical regions from public databases, center for disease control and prevention (CDC), and health authority reports to perform the analysis. For the SARS-CoV-2 virus driving COVID-19 pandemic, millions of genomes are available for investigation. One classical analytical method in the field of evolutionary biology is the phylogenetic tree (Fig 1), which is built upon clustering of viral strains according to their mutual distances. The tree provides an intuitive delineation of the ancestral relationship of viral strains and historical course of evolutionary pathway. Several tree-based methods have been proposed to predict future evolutionary trajectory by learning the dynamic pattern informed from the tree-clad (Luksza and Lassig 2014) or branching process (Neher et al 2014, Huddleston et al 2020). Because of the reliance on tree structure, predictions made by these methods fall on a cluster of strains. In our investigation of virus evolution, however, an alternative methodology is adopted. Instead of treating an entire sequence as the basic analytical unit, we consider site-wise evolution as the primary target of modelling. This approach was first motivated from our observation that key mutations appeared in stages through seasons – some advantageous mutations emerged years before an epidemic peak, and some followed in subsequent years and during the peak season (Wang, Lou et al 2021). Within individual host, billions of virions were replicated, carrying advantageous, deleterious, and neutral mutations. Under high pressure of host immunity, the virions carrying advantageous mutations enabling the virus to escape from immune recognition such as antibody binding would grow in larger quantities. Genetic variants carrying the leading mutations are more likely to be transmitted to other hosts. The subsequent mutations, also recognised as "trailing" or "permissive" mutations, play roles in maintaining viral functions and protein structure stability and facilitate the shaping of a predominant antigenic variant. Therefore, competition and selection of virus might start early with residue substitutions before the formation of predominant strains. Modelling site-wise evolution may gain lead-time to catch the large effect mutations that set the course of evolutionary pathway. We proposed several new quantities to characterise this process, including a time factor to describe the effective period of mutations to stay in-advantage against population immune response, and a threshold of mutation prevalence to signal when a mutant's impact in population epidemics is reaching an influential level. Based on these building blocks, we developed prediction model that projects genome-wide site-wise mutation dynamics to the next epidemic season, and through which to identify the optimal strains representing the future virus population that could be considered as vaccine viruses (Lou et al 2022). The model predicted vaccine strain, if manufactured as it is, is expected to improve the current vaccine's effectiveness by absolute 11.2% (95% CI: 3.5–18.8) against the H3N2. Prediction algorithm is also being developed for the SARS-CoV-2 virus evolution. Current data shows that it could capture the key mutations in the Omicron variants 6-12 months before it became predominant in population.
Predicting Vaccine Effectiveness
In early 2021, my husband asked, "What have you been working on lately?" I answered, "Something really exciting! We developed a method that can predict vaccine effectiveness (VE) before people taking the shot, and before a genetic variant is transmitted to a population." He said, "Alien thoughts!". In 2022, our research group published the world first method to predict COVID-19 VE by genome analysis, reaching 95% prediction accuracy (concordant correlation coefficient) in independent datasets (Cao et al 2022). Predicting VE by computational models can offer rapid assessment of vaccine protection facing emerging variants, while waiting for the golden standard measure of VE to be obtained from clinical trials or observational studies, which may take a long time spanning the administration of vaccines, population infection, clinical diagnosis, and data analysis. Using the model, government officials can be informed early of the threats on health system by new genetic variants and design timely public health response policy. Vaccine manufacturers can also design antigen and clinical trial according to estimated VE outcomes.
Behind the algorithm is a statistical model that connects the molecular level variations coming from virus mutations and the population-level VE. Sounds simple as it is, challenges exist for solving this problem. First, we need an informative metric that can capture the influential mutations on vaccine protection. For the influenza virus, this was tackled by evaluating genome loci that can best correspond to VE change, a process called feature selection in statistical learning. For the SARS-CoV-2 virus, we identified that the most informative genome region for VE is the receptor binding domain on the Spike protein, which is as a major target of host immune recognition. Another challenge is data quality, as the virus genome data and vaccine protection data are collected in separate studies and cohorts, we could at best match the two quantities according to their collection time periods and geographical regions. Due to sparsity of genome sequencing data and non-standardised VE studies in different countries, the best VE prediction model for the influenza virus had a R-squared of 55.0% for A H1N1pdm09 and 87.8% for H3N2 (Cao et al 2021, Cao et al 2022b). In 2021, COVID-19 VE reports and massive genome sequencing data of SARS-CoV-2 became available. We immediately extended the previous prediction model of influenza on COVID-19. A surprisingly accurate model can be built - benefit from the high-quality genome and VE data collected world-wide during the pandemic, the model's R-square reached 87.9%. Nevertheless, new challenge appeared. Different from the influenza vaccine that is mainly produced with a single platform using the inactivated technology, the COVID-19 VE possesses extra variations due to the diversified vaccine technology platforms, such as the mRNA, viral-vector, and protein subunit vaccines. New methods were introduced to handle it. Eventually, a highly precise VE prediction model is developed. For instance, we projected that the VE against symptomatic infection for Delta variant should be 82.8% by a mRNA vaccine, and the observed VE in real-world survey study was 83.0% (Fig 2).
These predictions methods comprise our many efforts toward transforming vaccinology using advanced bioinformatics and computational biology methods. Accurate in silico prediction and vaccine evaluation would enable design of vaccine antigen from a reverse vaccinology perspective - to inform the optimal practice now by predicting the future.
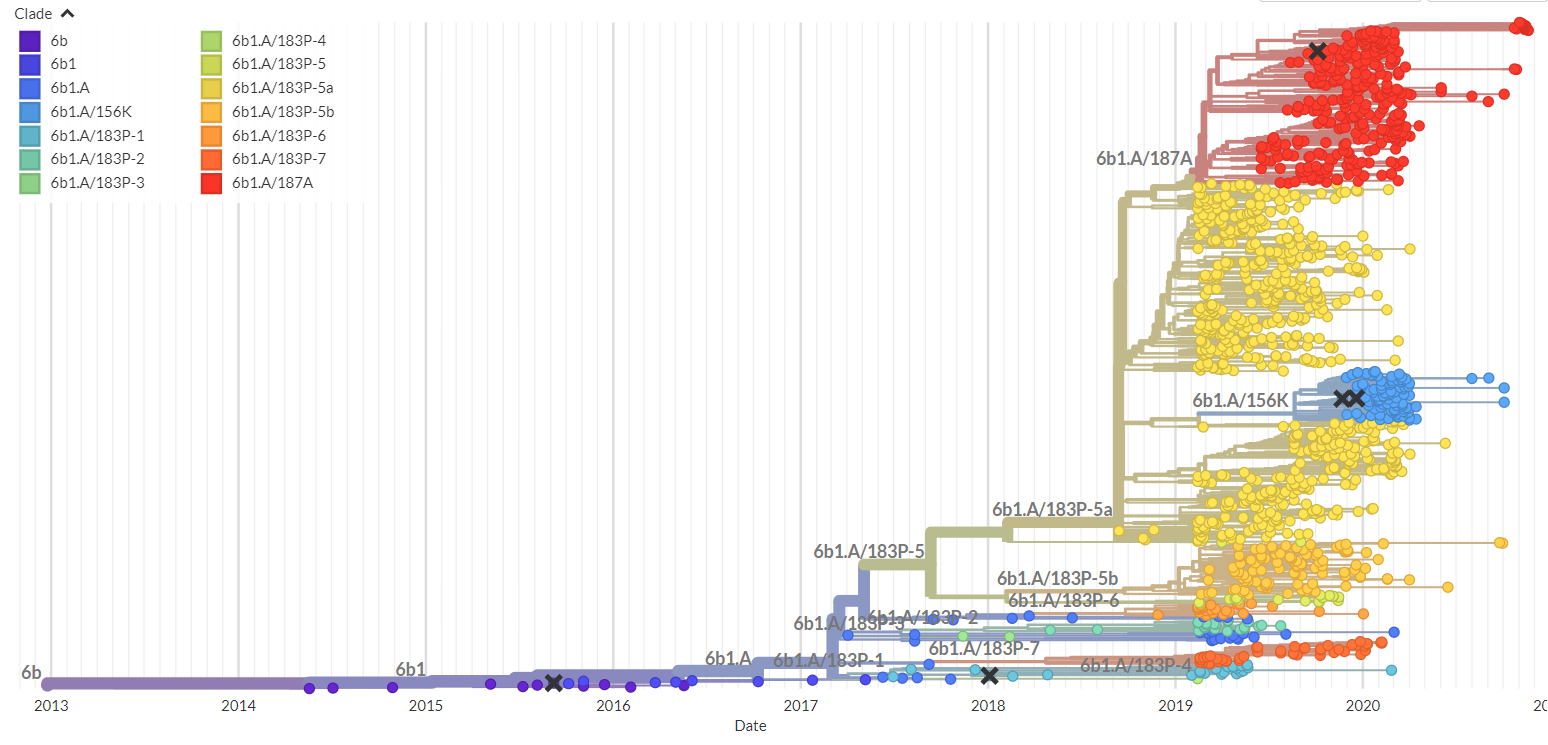
Fig 1: A phylogenetic tree for SARS-CoV-2 virus plotted by the Nextstrain (https://nextstrain.org/)
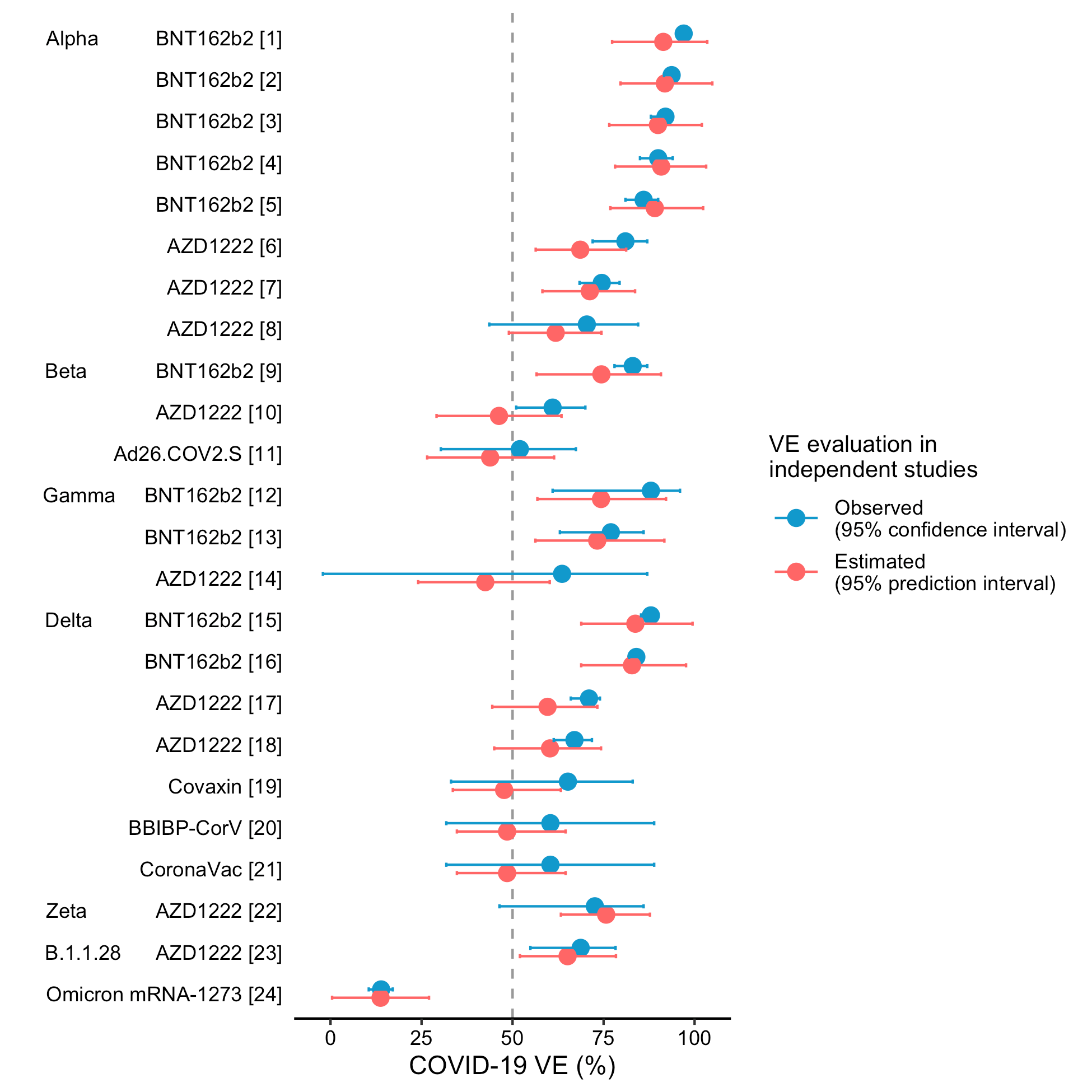
Fig 2: Predicted and observed vaccine effectiveness of COVID-19 vaccines (adopted from Cao et al 2022)
References:
- Cao, L. et al. Rapid evaluation of COVID-19 vaccine effectiveness against symptomatic infection with SARS-CoV-2 variants by analysis of genetic distance. Nat Med, doi:10.1038/s41591-022-01877-1 (2022).
- Cao, L. et al. In silico prediction of influenza vaccine effectiveness by sequence analysis. Vaccine 39, 1030-1034, doi:10.1016/j.vaccine.2021.01.006 (2021).
- Huddleston, J. et al. Integrating genotypes and phenotypes improves long-term forecasts of seasonal influenza A/H3N2 evolution. eLife 9, doi:10.7554/eLife.60067 (2020).
- Luksza, M. & Lassig, M. A predictive fitness model for influenza. Nature 507, 57-61, doi:10.1038/nature13087 (2014).
- Neher, R. A., Russell, C. A. & Shraiman, B. I. Predicting evolution from the shape of genealogical trees. eLife 3, doi:10.7554/eLife.03568 (2014).
- Wang, M. H. et al. Characterization of key amino acid substitutions and dynamics of the influenza virus H3N2 hemagglutinin. The Journal of infection 83, 671-677, doi:10.1016/j.jinf.2021.09.026 (2021).
Author:
Prof Maggie Wang, School of Public Health and Primary Care, The Chinese University of Hong Kong
January 2023