预测流感病毒的演化
我们经常听到有人问:基因突变不是随机的吗?如果是随机的,病毒演化可以预测吗?事实上,病毒演化不同于基因突变。突变是指病毒在复制过程中出现的随机性错误,它没有特定的方向。而病毒演化则是由病毒突变、宿主免疫反应、以及环境共同构成的一个复杂过程。因此,虽然病毒基因的突变难以捉摸,我们却可透过模型追踪宿主中优势突变的动态来预测病毒的短期演化。正如尽管我们不知道风从哪里来,但我们可通过观察受风影响的物体来捉摸风的方向和力量。同理,宿主的免疫反应和环境增大病毒基因改变的压力,其中一些关键的突变会体现出人体免疫反应的选择趋势和病毒的传播情况。因此,透过研究病毒与其宿主共同刻画出的历史数据,我们便能够了解病毒的演化特征。
铺垫了病毒演化预测的基本原理,建立病毒演化的模型仍是一份艰巨的工作,这需要收集大量的数据将病毒基因组和全球各地病毒流行的状况串联起来进行分析。比如,要捕捉H3N2病毒(季节性流感的其中一种主要亚型)演化的模式,我们要收集20 年间的基因数据以及十多个国家疾控中心(CDC)发表的流感监测数据,并从中学习。对新冠病毒,我们需要研究分析上百万个病毒基因组。在演化生物学中,根据物种基因组之间的相互距离对物种进行聚类而建立的系统发育树(图 1)是一种经典的分析工具,它能清晰的展示毒株之间的祖源关系以及病毒的演化历程。通过对系统发育树的分支优势(Luksza 及 Lassig,2014)或分支进程(Neher 等人 2014、Huddleston 等人,2020)进行建模,研究人员提出了几种方法来预测病毒未来演化的趋势。然而,我们采取了另一个角度来进行有关病毒演化的预测,不是如系统发育树般把整个基因序列当作基本分析的单位,而是把核苷酸位点上的演化动态视为模型要捕捉的主要目标。这种方法源于观察,即关键突变往往分阶段发生,部分有利的突变在疫情爆发高峰前几年已经出现,其余的则在随后几年和高峰季节期间发生(Wang、Lou 等人,2021)。在宿主个体内,数十亿计的病毒被复制,产生有利、有害和中性的突变。在宿主免疫的高度压力下,携带有利突变的病毒能避开如抗体识别的免疫机制,并大量复制。那些携带重要突变的病毒更有机会传播给其他宿主,其后产生的突变(亦称为「尾随」或「辅助」突变)常常具有维持病毒的功能或稳定其蛋白质结构的功能,促进主流变种病毒的形成。因此,在主流毒株形成以先,病毒已经在残基置换下开始进行竞争和选择。这表示,建立基于核苷酸位点的演化模型可能会让我们更早找到影响演化历程的重要突变。我们提出了几个新的定量来量化这个过程,其中包括突变的有效期,它描述新变异在人群免疫反应下保持优势的时间;以及突变爆发的门阀值,它描述突变体的适应性到达何种程度时该病毒会引发新一波的大量人群感染。基于这些以及其他信息,我们开发了一套预测模型beth-1,找出能够代表未来病毒群组的最佳毒株,以作为制作疫苗的抗原蓝本(Lou 等人,2022)。该模型所预测的疫苗株(如按设计生产),预计可使现有疫苗对 H3N2 的保护效力升高绝对11.2%(95% CI:3.5-18.8)。我们亦正为 SARS-CoV-2 病毒的演化开发预测演算法,目前数据显示,该模型能够在 Omicron 变种病毒在人群中成为主流病毒的 6-12 个月前,找到关键突变。
预测疫苗的效力
在2021 年初,先生问我:「最近在忙些什么?」答:「让人非常振奋的工作!我们找到一种方法可以在人们接种疫苗或感染病毒以前,预测疫苗效力。」他道:「匪夷所思!」2022 年,我们研究小组发表了全球首个透过基因组分析来预测新冠疫苗效力的方法(VE-GD 模型),在独立的数据组中预测准确度(一致相关系数)达到 95%(Cao 等人,2022)。如要从临床试验或观察性研究中取得疫苗效力的黄金标准测量值,通常需要较长的时间来进行疫苗接种、人群感染、临床诊断和数据分析。如果可以用电脑模型来估算疫苗效力,便可迅速评估疫苗在新变种病毒挑战下的保护性,令政府官员尽早知悉新变种病毒对卫生系统的威胁,以及时制定适当的公共卫生政策。此外,疫苗生产厰亦可根据疫苗效力的预估结果,设计出更优化的抗原和临床试验。
这个VE-GD演算法是通过统计模型实现的,它将病毒突变所引起的分子水平变化以及疫苗在人群层面所产生的效力连系起来。虽然听起来很简单,但要完成这个设想却并不容易。首先,我们需要找出对疫苗保护力有影响的突变。在对流感疫苗的研究中,我们通过统计或机器学习中称为特征选择的方法来解决这个问题。在新冠疫苗上,我们发现对疫苗效力影响最大的基因组区域是刺突蛋白上的受体结合区,它正是宿主免疫识别的主要目标。另一个研究的难处是疫苗数据的质量,由于各国对流感病毒基因组的测序数据零散稀少,疫苗有效性的研究欠缺标准,因此疫苗效力预测模型对流感病毒 A /H1N1pdm09 的最佳表现仅为55.0%(R方),对 H3N2为 87.8%(Cao 等人,2021、Cao 等人,2022b)。在2021 年,随著大量关于新冠疫苗有效性的优质报告的出现,模型的准确性得到了大幅上升。然而,新的挑战出现了。不同于流感疫苗采取相当单一的技术,新冠疫苗具备多种疫苗技术平台,例如 mRNA、病毒载体,以及蛋白亚单位疫苗,因此疫苗有效性出现了更多变化。于是我们引入了新的方法来解决这个挑战,最终建立了一个相当精准的预测模型。比如,该模型预测 mRNA 疫苗针对 Delta 变种病毒有症状感染的疫苗有效性为 82.8%,在实际数据中得到的疫苗效力为 83.0%(图 2)。
这些预测方法构成了我们在生物科技领域许多努力中的一部分,即通过生物信息学以及统计学方法助力疫苗研发。精准的计算和预测疫苗抗原表现,能够帮助我们从反向疫苗学的角度设计出更优越的疫苗抗原 —— 预测未来,以引导当下。
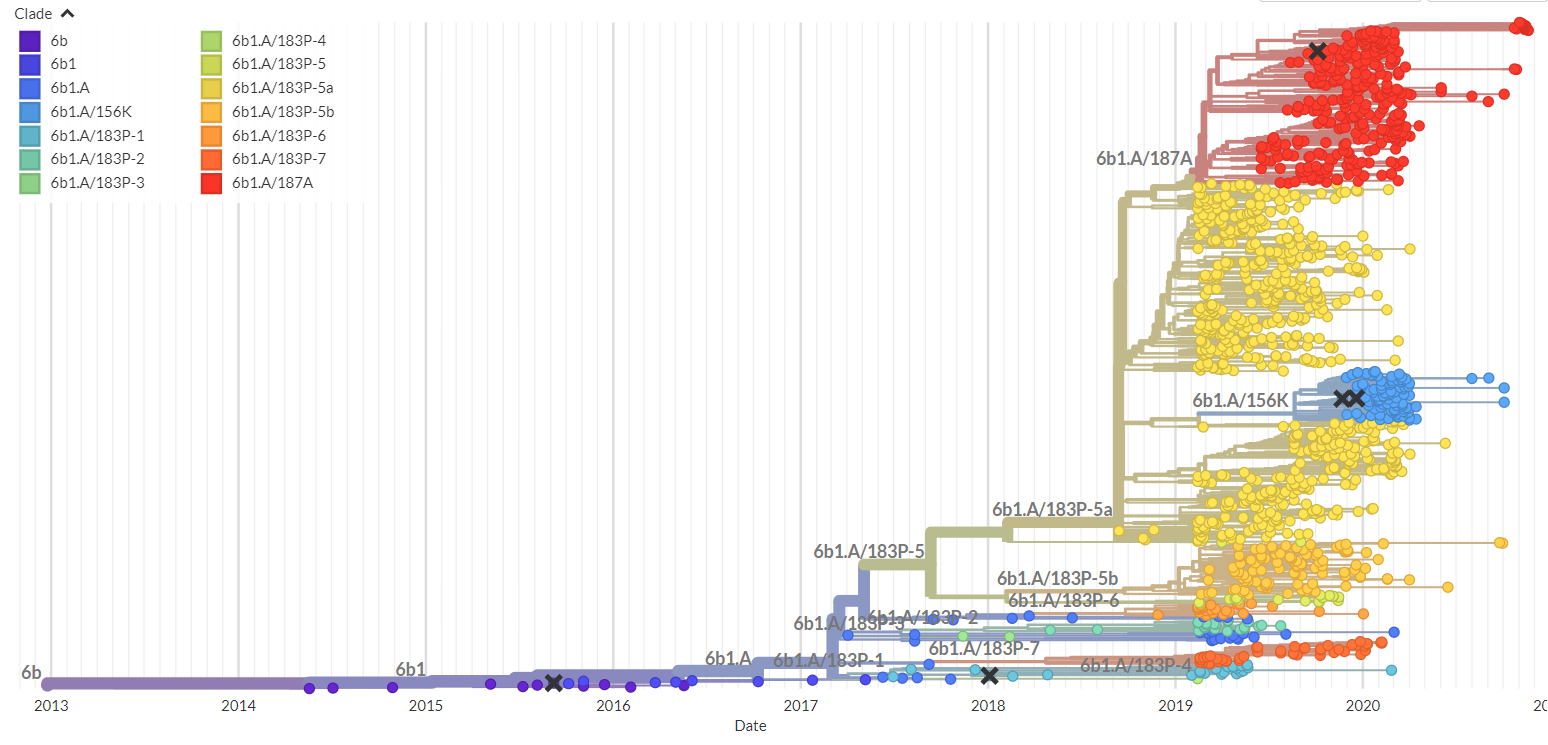
图1: 由Nextstrain绘制的SARS-CoV-2病毒系统发育树 (https://nextstrain.org/)
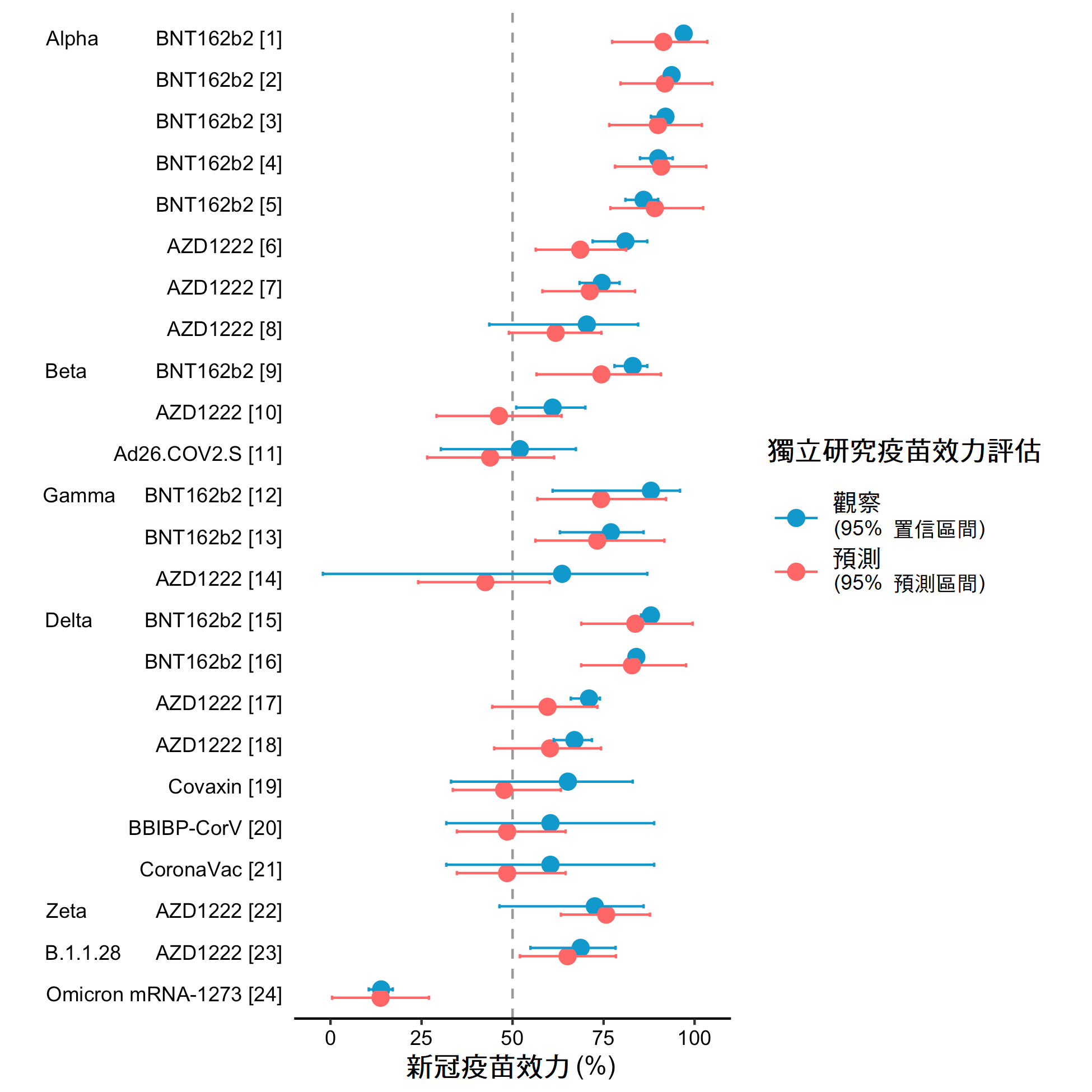
图2: 预测及观察新冠疫苗的疫苖效力 (来源︰Cao等人, 2022)
参考资料︰
- Cao, L. et al. Rapid evaluation of COVID-19 vaccine effectiveness against symptomatic infection with SARS-CoV-2 variants by analysis of genetic distance. Nat Med, doi:10.1038/s41591-022-01877-1 (2022).
- Cao, L. et al. In silico prediction of influenza vaccine effectiveness by sequence analysis. Vaccine 39, 1030-1034, doi:10.1016/j.vaccine.2021.01.006 (2021).
- Huddleston, J. et al. Integrating genotypes and phenotypes improves long-term forecasts of seasonal influenza A/H3N2 evolution. eLife 9, doi:10.7554/eLife.60067 (2020).
- Luksza, M. & Lassig, M. A predictive fitness model for influenza. Nature 507, 57-61, doi:10.1038/nature13087 (2014).
- Neher, R. A., Russell, C. A. & Shraiman, B. I. Predicting evolution from the shape of genealogical trees. eLife 3, doi:10.7554/eLife.03568 (2014).
- Wang, M. H. et al. Characterization of key amino acid substitutions and dynamics of the influenza virus H3N2 hemagglutinin. The Journal of infection 83, 671-677, doi:10.1016/j.jinf.2021.09.026 (2021).
作者︰
香港中文大学公共卫生及基层医疗学院王海天教授
2023年1月