簡介
這個故事的主角是2007年邵逸夫生命科學與醫學獎得主羅伯特‧尼科威教授的得獎研究,G蛋白偶聯受體(G Protein-Coupled Receptors, GPCR)蛋白質家族的其中一個成員。尼科威教授後來亦因同一研究課題於2012年與布萊恩‧科比爾卡教授共同獲得諾貝爾化學獎。GPCR是介導細胞及器官對藥物和激素反應的主要受體系統。
故事源起於在一群性早熟男童裏發現一種名為「家族性男性性早熟」(familial male-limited precocious puberty, FMPP)、具100%外顯率的常染色體顯性遺傳病。患病的男童早至2至4歲就出現青春期特徵,原因是其體內的男性激素睾酮達到成年男性的水平。而黃體生成激素(luteinising hormone, LH)量僅達青春期前的水平(LH是青春期時調節睾酮分泌量的激素)。由於異常情況是由睾酮過量分泌所導致,因此臨床研究最初亦以睾酮中毒症(testotoxicosis)來描述此情況。由於已得知細胞表面受體——LH受體(LHR)會介導在睾酮分泌時LH的活動,因此研究人員檢查了FMPP患者體內的受體,並發現LH受體出現活化突變。而研究亦意外發現FMPP患者患有另一種相對罕見的病症——睾丸腫瘤,以致亦發現了會引致腫瘤的LHR突變分支。這項發現改變了FMPP患者的傳統臨床治療法。這是本次研究剛開始時,完全未預料到的結果。
黃體生成素/絨毛膜促性腺激素受體(Luteinising Hormone / Chorionic Gonadotropin Receptor, LHR)
LHR是GPCR家族的成員。這個蛋白質家族的成員一般有七個跨膜(transmembrane , TM)螺旋,這些螺旋會將與蛋白質胞外區結合的藥物或激素的信號轉導至細胞內的第二信使系統。激素與受體的結合會活化受體,引發一連串膜和細胞內活動。LHR有一個長N端細胞外區、七個跨膜α螺旋及一個短C端細胞內區1。人類LHR(hLHR)基因的編碼區由10個內含子及11個外顯子組成。前10個外顯子為大部分細胞外區編碼;外顯子11則只為小部分N端細胞外區、全部7個TM區以及C端細胞內區編碼。細胞外區乃負責LH的高親和力結合。不同物種的TM區(包括TM螺旋及連接環)都高度一致,而TM區亦與其他糖激素受體高度一致。這個區以及胞質C端區對於轉導引發第二信使系統活化的激素結合信號(腺苷酸環化酶級聯反應)非常重要,會導致cAMP的合成增加,以及啟動一連串膜和細胞內活動。而所引致的基因活化會增加睾酮分泌以及雄性性發育。由於同一受體還會與人類絨毛膜促性腺激素(human Chorionic Gonadotropin, hCG)結合,因此又稱為黃體生成素/絨毛膜促性腺激素受體(Luteinising Hormone / Chorionic Gonadotropin Receptor, LH/hCGR,或簡稱hLHR)。大多數由LH信號異常引發的人類疾病都是hLHR突變的結果。
LHR的功能獲得型活化突變–家族性男性性早熟(LHR—Familial Male-limited Precocious Puberty, FMPP)
人類的睾酮幾乎全部由睾丸產生(超過95%)。正常生理條件之下,睾酮的主要來源是睾丸內的睾丸間質細胞。睾丸間質細胞表面的跨膜hLHR會介導LH的作用。由於hLHR在LH的作用下會觸發睾酮的合成,因此我們先檢查FMPP患者的hLHR。這個步驟找出了hLHR的單點活化突變,並確定了患者患上該病的病因。
FMPP是由活化突變所引致,而這些突變屬功能獲得型突變,並會引起男童的非性腺釋素(luteinising-hormone releasing hormone, LHRH)依賴型同性早熟。hLHR的組成型活性會在無激素的情況下刺激胎兒時及青春期前的睾丸間質細胞,導致睾酮自行分泌。患病的男童最早會在3至4歲出現第二性特徵、陰莖生長、雙睾增大、陰毛生出,與真正的青春期無異。以往沿用的診斷法是量度睪酮水平。雖然患病男童的促性腺激素符合該年齡段的基礎水平(即青春期前水平),但是他的睪酮卻達到青春期至成年期的睾酮水平。該病之所以曾經名為睾酮中毒症,是因為當初在青春期前的患者身上測到達中毒水平的睾酮。如進行睾丸活體組織學檢查,會發現睾丸間質細胞有生長2。hLHR的活化突變對女性患者無可見影響。治療這種病可使用兩種藥物:螺內酯(spironolactone,抗雄激素藥,能阻止睾酮起作用),以及睾內酯(testoleactone,抗芳香酶,能阻止睾酮形成)。進入青春期後,LH分泌會激增,觸發睾酮的產生以及男性性發育,因此到青春期時會停用這兩種藥,以便天然產生的睾酮起作用。因此,當睾酮中毒患者進入自然青春期,一般均不會繼續追縱。
FMPP的分子診斷
所有已知的FMPP突變都是在hLHR基因的外顯子11(負責為TM區編碼)上發現的單點錯義突變。由於所有已知的hLHR活化突變都位於一個大外顯子上,換言之可以利用聚合酶連鎖反應(Polymerase chain reaction, PCR)進行DNA擴增然後測序,這種FMPP分子診斷方法會較為直接簡單。在文獻報導的120多名FMPP患者之中,超過50%是經由本人的實驗室所確診的。研究發現患者在15種氨基酸上出現18種不同的突變(圖1A)3,4。一種氨基酸殘基(Asp578)有3種不同的突變;另一種氨基酸殘基(Asp564)則有2種不同的突變。約10%的突變是偶發突變(即新突變),而在FMPP家族中,有10%並無可見的hLHR異常狀況。大多數FMPP突變出現在TM VI,因此TM VI是突變的熱點。最常見的突變氨基酸是Asp-578。在所有活化突變之中,超過55%有影響該氨基酸。至於受家族及受新突變影響的患者,兩者之間未見有臨床表現上的差異。在非高加索族裔背景的患者,包括西班牙裔人、亞洲裔人、非洲裔美國人、非洲裔巴西人之中,稀有突變的發生率較高3。
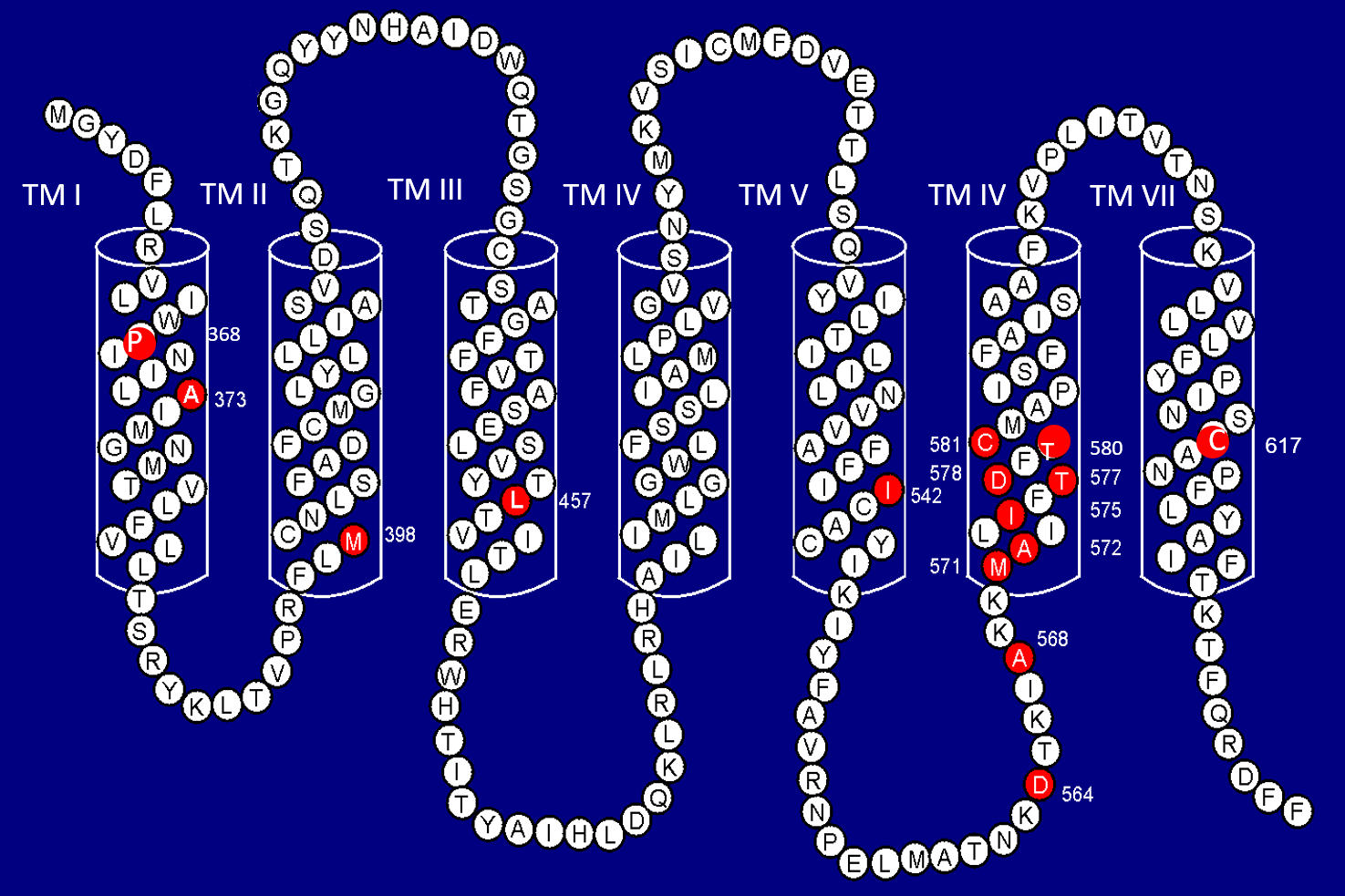
圖1A﹕人類黃體生成素/絨毛膜促性腺激素受體(Human Luteinizing Hormone/Chorionic Gonadotropin Receptor, LHR)。
七個跨膜螺旋(TM I至TM VII) 紅色實心圓圈代表在FMPP患者體內發現的突變氨基酸殘基。氨基酸殘基位置以數字表示。
由診斷至分子機制
人類黃體生成素受體(human luteinising hormone receptor, hLHR)以功能獲得型突變活化,其機制大體上不明。我們調查hLHR的致病突變以及去活化突變後,得以利用結果進行hLHR的分子模型研究。大多數活化突變見於TM螺旋,尤其是hLHR的螺旋VI。進行分子模型研究,以及將TM VI與突變氨基酸並排作序列對比後,結果顯示,受hLHR致病突變影響的氨基酸乃位於螺旋束的內部,指向由七個TM螺旋形成的導管之內(見圖1B及表1)。對FMPP患者的Asp-578自然突變及定點誘變的研究顯示,由Asp-578突變引起的hLHR活化,其活化程度大體上取決於更代氨基酸的側鏈大小,而非其電荷5。因此,這些氨基酸的突變將改變螺旋間相互作用以及導致活化的受體的構形, 而這種改變可能實際涉及由螺旋形成的導管的張開。
因此得出結論﹕Asp-578在hLHR的信號轉導中擔當位置恰當的氫鍵受體,發揮重要作用。我們對位於螺旋之外的氨基酸殘基進行了定點誘變研究,結果顯示其他類型的相互作用(例如靜電及空間相互作用)對於受體的活化亦可能有重要作用5。
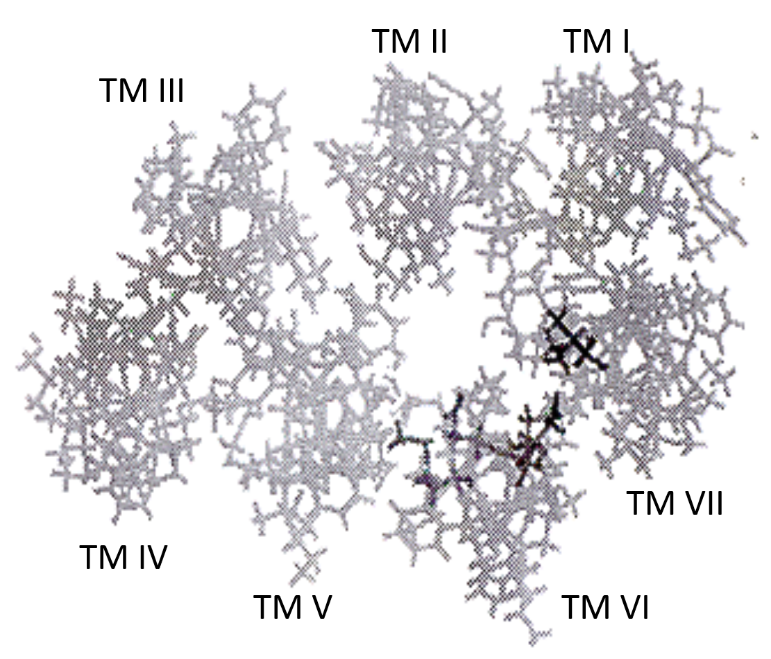
圖1B﹕人類黃體生成素/絨毛膜促性腺激素受體(Human Luteinizing Hormone/Chorionic Gonadotropin Receptor, LHR)。
跨膜螺旋電腦模型鳥瞰圖。較深色符號代表突變氨基酸殘基。
| 正常 AA | 突變 | 位置 # | 螺旋轉角 # (3.6 AA/轉角) |
疾症 |
|---|---|---|---|---|
| Met-571 | Ile | 1 | 0 | FMPP |
| Ala-572 | Val | 2 | 0 | FMPP |
| Ile -575 | Leu | 5 | 1 | FMPP |
| Thr-577 | Ile | 7 | 2 | FMPP |
| Asp-578 | Gly/Tyr/Glu | 8 | 3 | FMPP |
| Cys-581 | Arg | 11 | 4 | FMPP |
| Ile-585 | No known | 14 | 5 | - |
| Ala-589 | No known | 19 | 6 | - |
| Ala-593 | Pro | 23 | 7 | LCH |
表1﹕TM VI內各種突變氨基酸殘基(AA)的並列比較。
並無檢測到氨基酸殘基Ile-585及Ala-589的突變。
另一個極端——功能喪失型失活突變——睾丸間質細胞發育不良(Leydig Cell Hypolasia, LCH)
另一方面,類似的錯義突變亦可使受體失活,導致男性性發育低於正常水平。失活突變屬功能喪失型突變,會導致受體無法與激素結合或已結合的激素無法活化受體。這類突變會影響性發育,特別是對男性。當男性體內發生純合子或複合雜合子失活突變時,hLHR活性的降低或廢止會導致對LH刺激產生抗性,令睾丸間質細胞無法分化,於是出現FMPP的相反——睾丸間質細胞發育不良(LCH)或睾丸間質細胞不發育(Leydig Cell Agenesis),是假性雌雄同體(pseudohermaphroditism)之一種。LCH的臨床表現並不一致。一邊極端是遺傳上是男性但長有外觀正常的女性外生殖器,他們最後往往會接受變女性手術。另一邊極端的個案則是患有高促性腺素性功能减退症(hypergonadotropic hypogonadism)、小陰莖以及男性外生殖器發育不良。而兩者之間的個案則是程度各異的外生殖器男性化。由於任何突變都有可能使已編碼的蛋白質失活,因此LCH的分子遺傳學表現比FMPP更為不一。在LCH患者中發現的hLHR失活突變包括錯義突變、無義突變、插入、缺失。另外在若干LCH個案中並未發現有hLHR突變,表示失活突變的樣式繁多,或者LCH是由hLHR以及其他基因的突變所引起。我們所觀察到的遺傳突變有多種形態表現,同樣,各種突變對hLHR作用的影響亦各有不同。視乎受體蛋白中突變位置的不同,突變可導致激素的結合減少、表面表現減少、細胞內轉運異常,或偶聯效率降低,所有情況都會導致激素結合所觸發的信號轉導減少或缺失6。
新的波折——癌症中的生殖系突變及體細胞活化突變
一般而言,當對某疾病作分子遺傳學研究時,能設計出診斷方法以及解構出疾病的機制已可以稱得上是達到研究的目標了。然而,FMPP的研究卻出現另一番波折。有一位患者在27個月大時在喬治城大學醫學中心根據內分泌評估被診斷患有FMPP,後來到35歲時獲診斷出有睾丸精原細胞瘤。為確認該患者的FMPP診斷,他接受了對hLHR基因的分子分析,並發現體內有活化突變Asp578Gly。這是首宗確診FMPP患者患上睾丸腫瘤的報告7。在此報告之後,又發現另一名FMPP患者出現活化突變Asp564Gly,而且長有兩個無症狀睾丸間質細胞瘤8。由於突變是在循環血細胞基因組DNA中檢測得到,因此兩個個案的hLHR突變都屬生殖系。在第一位FMPP患者被診斷出有睾丸精原細胞瘤一年後,Liu等人9報告有三名患者在睾丸間質細胞腺瘤組織內的hLHR基因中出現活化突變Asp578His。該項突變並未在相鄰的正常組織或血細胞之中出現。因此,儘管未知高水平的睾酮產生會否導致腫瘤形成,但是具有組成性活性的hLHR或會使人易患睾丸腫瘤。醫學界以往會在FMPP患者進入自然青春期後送其出院,這項發現改變了傳統的做法,並提出有必要在患者進入青春期之後繼續追踪,檢查他們是否出現早期睾丸腫瘤。
hLHR的突變偏向信號途徑
研究hLHR突變影響的細胞模型
我們在FMPP患者中從未檢測到Asp578His突變;這種突變乃體細胞突變,不會經生殖細胞傳遞至下一代。Asp578His的致癌機制尚未得知。以上觀察結果為hLHR活化突變的生物效應研究開闢了新的方向。
為探究活化突變的遺傳效應,我們建立了一套細胞模型。我們利用老鼠睾丸間質細胞系MA-10細胞(這細胞還保有睾丸間質細胞部分生成類固醇及對LH的刺激有反應的性質)來比較Asp578Gly與Asp578His突變對睾丸間質細胞的影響。我們使用了野生型hLHR(hLHR-WT)的重組pcDNA3質體、帶有Asp578Gly(hLHR-D578G)或Asp578H(hLHR-D578H)的hLHR來轉染MA-10細胞。我們使用了老鼠全基因組晶片來檢查三種細胞的表達譜,即MA-10W(表達hLHR-WT的MA-10細胞)、MA-10G(表達hLHR-D578G的MA-10細胞)及MA-10H(表達hLHR-D578H的MA-10細胞)。如圖2A所示,我們用無引導階層式分群法分析了野生型及突變型轉染子(每個類別3個複製)所表達的基因。分群法將表達譜明顯分成三組,與所表現的hLHR基因型一致。MA-10G及MA-10H與對照的MA-10W細胞迥然不同。相比MA-10W,MA-10G及-10H有132個調升的基因。其中大多數是癌基因以及與發育(特別是神經元發育)有關的基因。我們對MA-10W與兩個突變細胞(MA-10G及MA-10H)之間表達差異的基因進行了本體分析,發現兩個突變體有共同的七個生物學類別(表2)。然而,兩類在MA-10G細胞發現的基因(即與發育成熟及繁殖有關的基因)在MA-10H突變體中並無出現, 因此確證了Asp578His突變的體細胞特性(該特性在FMPP患者中尚未發現)。
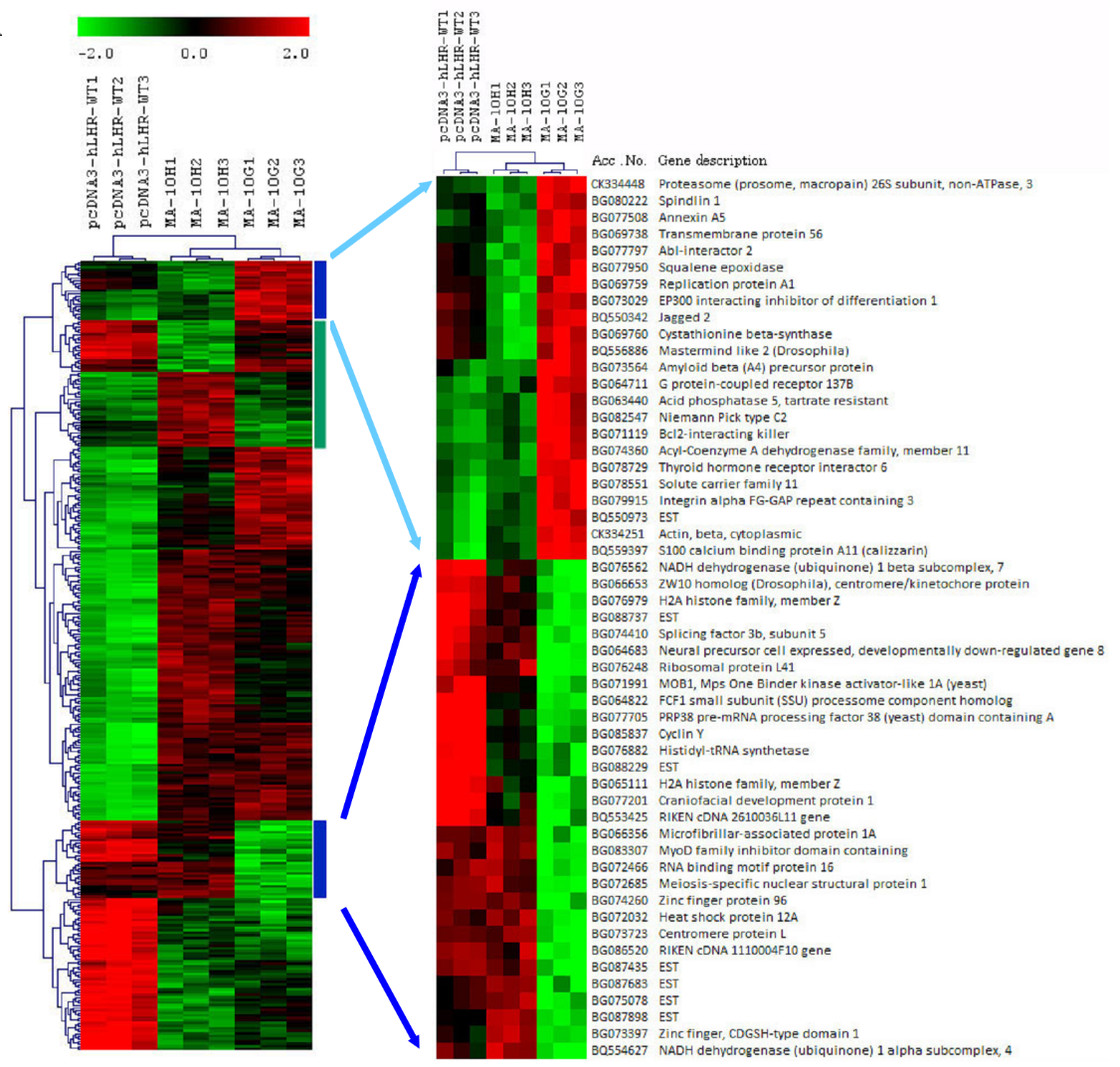
圖2A﹕MA-10轉染子的階層式分群法及突變專屬網絡的鑑認。
MA-10W(pcDNA3-hLHR-WT,複製1至3)、MA-10H(hLHR-D578H,複製1至3)及MA-10G(hLHR-D578G,複製1至3)裏的已表達基因階層分群 。藍色直線代表帶有與MA-10G細胞相關的表達特徵的基因群;綠色直線代表帶有與MA-10H細胞相關的表達特徵的基因群。為顯示基因名稱,特別放大藍線簇群。
| hLHR-Asp578Gly (MA-10G) | hLHR-Asp578His (MA-10H) |
|---|---|
| 細胞代謝過程 | 細胞代謝過程 |
| 發育 | 發育 |
| 生長 | 生長 |
| 生物之間的相互作用 | 生物之間的相互作用 |
| 生理過程 | 生理過程 |
| 生物過程調節 | 生物過程調節 |
| 對刺激的反應 | 對刺激的反應 |
| 發育成熟 | - |
| 繁殖 | - |
表2﹕MA-10G及MA-10H細胞根據基因本體分析表達譜的主要生物學類別分析
當比較MA-10G與MA-10H細胞的表達譜時,會發現在兩個突變細胞之間表現相異的兩個特徵基因群(圖2A,藍色條及綠色條),表示該兩個突變hLHR的基因組效應並不相同。由此可見,用兩種不同的氨基酸替換同一種氨基酸,會對相關基因的表達產生不同影響,是為hLHR裏有突變偏向基因表達的第一項證據。
我們試圖透過重新生成由基因群代表的細胞網絡來分辨與MA-10G及MA-10H突變體有關的網絡。為了在生物網絡的環境裏將基因表達資料視覺化,我們利用了由多個來源集合而成的GeneGo MetaCore資料庫分析基因群,以便查詢及檢索生物學感興趣的基因與蛋白質之間關係,並在集成網絡上獲得使生物學假設成立的拓撲不變量(topological property)。上述分析產生了分別與MA-10G細胞及MA-10H細胞相關的9及12個網絡。為了減低兩個突變體共佔融合網絡的可能性,我們消除了兩個網絡共有的所有基因元素。該過程分別產生了兩個及三個MA-10G細胞及MA-10H細胞專屬的網絡(圖2B)(表3)。突變專屬的網絡只涉及調升的基因。
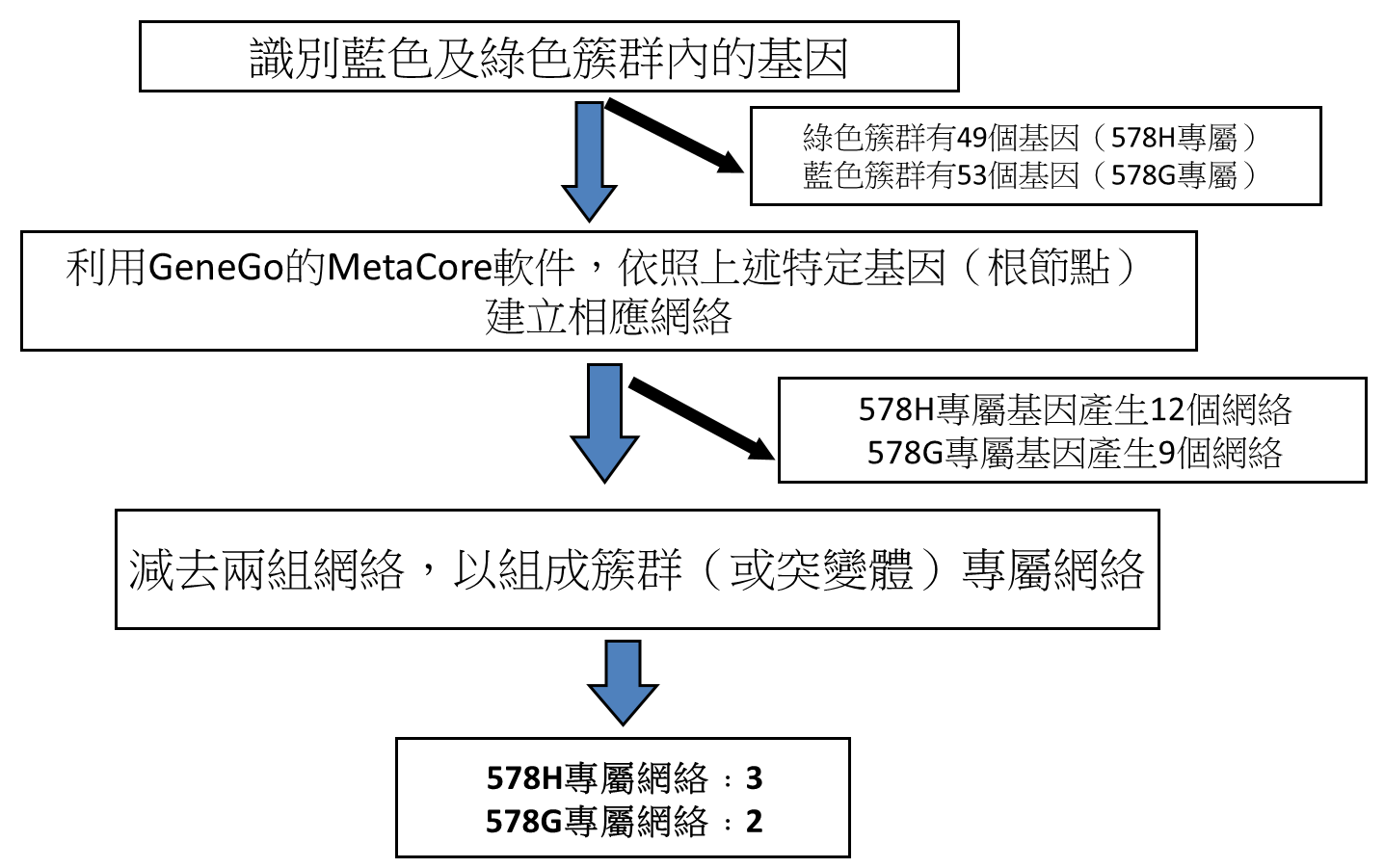
圖2B﹕MA-10轉染子的階層式分群法及突變專屬網絡的鑑認。
GeneGo的MetaCore分析,用作識別突變專屬網絡。
| 突變基因類型 | 網絡 | (調節子) 根節點 |
|---|---|---|
| Asp578Gly | 1 | (c-Myc) APP、Jagged2、CFDP1、H2AFZ、CBS |
| 2 | (PKC-alpha) APP-C59,膜聯蛋白 | |
| Asp578His | 1 | (c-Src)GUCY1A3,TRIF(TICAM1),Survivin |
| 2 | (nAChR)AARS,鳥苷酸環化酶,鈣調蛋白 | |
| 3 | (ESR1) MIF,轉移蛋白,C8orf40,腎上腺髓質素 |
表3﹕發現突變專屬網絡。
生殖細胞hLHR-Asp578Gly專屬信號網絡的確認
為了確認MA-10G或MA-10H細胞內有網絡存在,我們使用了siRNA來檢查關鍵成員(調節器)對網絡裏其他根節點以及對相應突變細胞之增殖的消除效果。舉例而言,我們的研究結果顯示,在兩個MA-10G專屬網絡之一裏,c-Myc(一種在控制細胞增殖、分化、凋亡及維持細胞內平衡中發揮重要作用的轉錄因子)可以利用與發育相關的多種功能(例如β類澱粉蛋白(A4)前體蛋白(App)、顱面發育蛋白1(Cfdp1)及胱硫醚β合酶(Cbs)來調節下游基因的表達,以及利用H2A組蛋白家族(H2afz)來調節對DNA結構完整度或基因活化10的維持(圖3A)。相比MA-10W(56%)及MA-10H(64%)細胞,MA-10G表現出更高的基礎c-Myc活性,並且對c-Myc siRNA抑制更敏感,因為其活性降低達40%(p < 0.05)(圖3B)。如表達譜分析所顯示,c-Myc是由Notch1調節;Notch1則是Jagged2的一個下游標靶;Jagged2則是一個被hLHR-D578G上調的基因。蛋白轉漬法(Western blotting)的結果證實了Jagged2對Notch1的調節;當中可見,用Jagged2 siRNA處理後,MA-10G細胞裏的Notch1裂解水平降低,Notch1受到抑制;但在MA-10H細胞裏,由於正常情況下Notch1蛋白量很少,因此並未明顯受抑制(圖3C)。實驗證實了c-Myc網絡在MA-10G細胞裏的活性,因為 c-Myc的消除可導致c-Myc網絡的所有組成部分(即App、Cfdp1、H2afz及Cbs)的表達被抑制(圖3D)。圖3E亦顯示,MA-10G細胞的增殖對c-Myc siRNA比對c-Src siRNA(對照)更為敏感,即是c-Myc網絡在該等細胞中更為重要。總括而言,以上實驗顯示,hLHR-D578G的專屬網絡之一是透過生殖系活化突變來上調Jagged2,此乃Notch1 – c-Myc調節作用的開關,並導致細胞增殖增加。
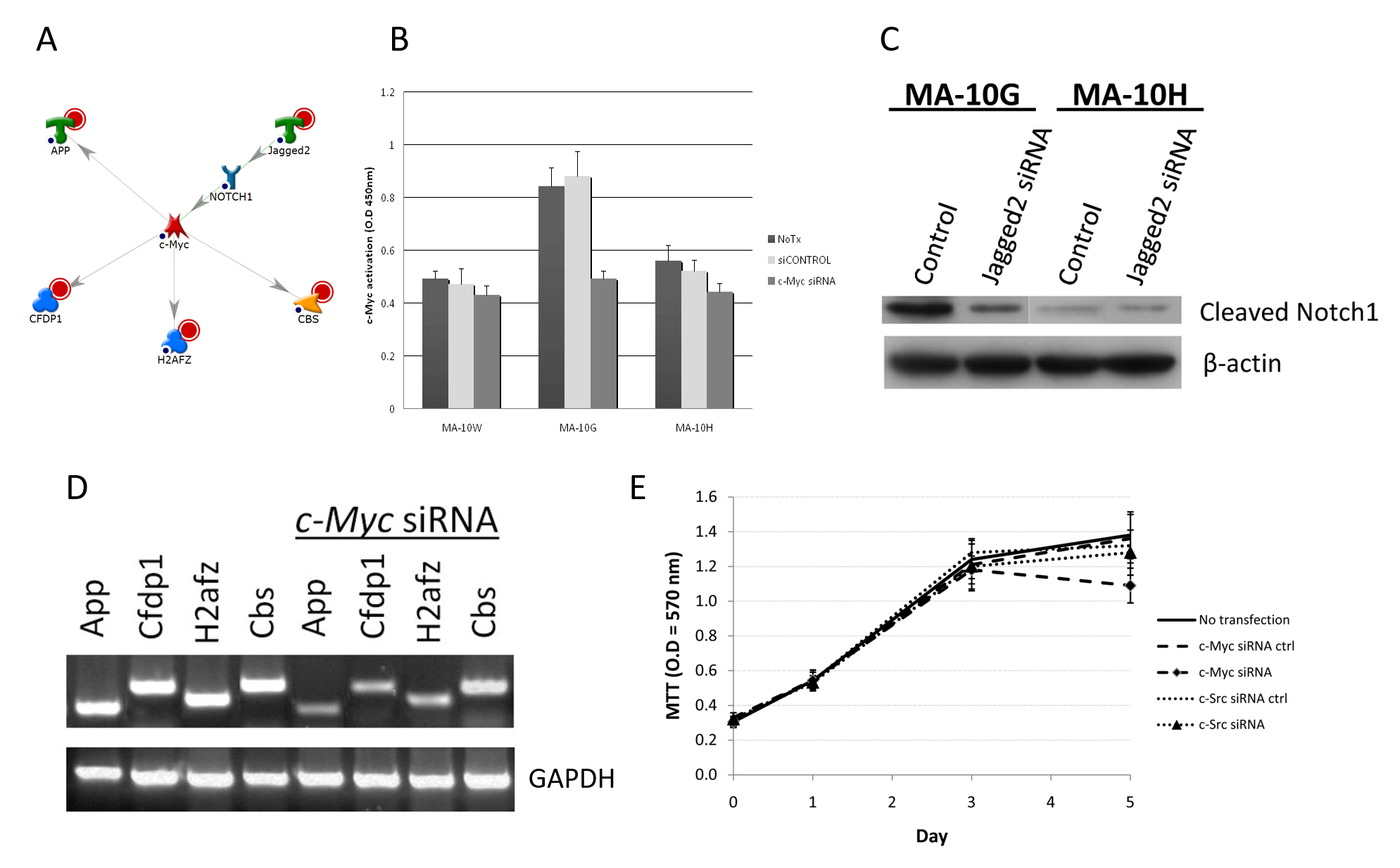
圖3A-E﹕生殖系hLHR-Asp578Gly專屬c-Myc信號網絡的確認。
A:MA-10G細胞內c-Myc網絡。紅色圓圈代表根節點。
B:c-Myc siRNA對MA-10G細胞內c-Myc活性的影響。根據廠商指引,短暫轉染100nM ON-TARGETplus siRNA(Dharmacon,Chicago,IL)24小時來抑制c-Myc及c-Src。為了確認個別siRNA在消除時的特異性,每次轉染實驗都設有ON-TARGETplus siCONTROL非標靶對照(Dharmacon,Chicago,IL)。使用TransAM c-Myc及FACE c-Src試劑盒(Active Motif,Carlsbad,CA)為活化的c-Myc及c-Src信號蛋白作定量。利用分光光度計將光密度定為450 nM,透過比色反應量度活化信號。MA-10G表現出較高的基礎c-Myc活性,並對c-Myc的抑製作用更為敏感,因為其活性降低達40%(p < 0.05)。
C:以Notch1為介體之Jagged2對c-Myc調節的確認。利用GO及網絡分析檢查MA-10G細胞內Jagged2調節c-Myc之時Notch1的參與。配體結合後,Notch受體被裂解,並釋放出一種細胞內亞基;該亞基遷移至細胞核,並在細胞核內擔當轉錄輔激活蛋白的作用。因此,我們以蛋白轉漬法檢查了MA-10G及MA-10H細胞內有或無Jagged2抑制作用時的Notch1裂解量。在MA-10G細胞內觀察到裂解的Notch1明顯減少(MA-10H則無)。
D:c-Myc對下游基因的調節。我們用c-Myc siRNA轉染細胞,然後進行半定量RT-PCR。我們在siRNA轉染前後均檢查了App、Cfdp1、H2afz及Cbs的表現。c-Myc的消除導致pp、Cfdp1、H2afz及Cbs的表達被抑制,可見MA-10G內c-Myc網絡的所有組成部分均有運行。
E:為了研究野生型或突變型hLHR對c-Myc的細胞增殖的特定作用,我們利用c-Myc siRNA轉染進行了五日的3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四唑溴化物(MTT)化驗。MA-10G的增殖對c-Myc siRNA的敏感度比對c-Src siRNA(對照)高,顯示c-Myc途徑在MA-10G細胞內更為重要。
體細胞hLHR-Asp578His專屬信號網絡的確認
在三個MA-10H專屬網絡之一裏,H專屬簇基因鳥苷酸環化酶1 alpha 3(Gucy1a3)、類鐸受體銜接子分子(Ticam1)及生存素(Birc5)是透過c-Src的控制而受調節(c-Src是一種已知的G蛋白受體標靶以及轉錄因子NF-κB的調節子)(圖4A)。C-Src(原癌基因酪氨酸蛋白激酶Src)是一種脂蛋白信號分子(經荳蔻酰化),透過多種底物的磷酸化參與細胞間相互作用、細胞遷移及增殖11。至於MA-10H細胞,研究發現其c-Src活性比MA-10W及MA-10G高,分別高26%及27%(圖4B)。儘管利用c-Src siRNA作轉染導致了MA-10G細胞裏的c-Src活性降低,但MA-10H細胞對相同的處理更為敏感,活性降低達33%(P < 0.05)。這項結果與先前對c-Myc的研究共同證實了c-Myc在MA-10G以及c-Src在MA-10H細胞中的主要調節作用(圖4B)。MA-10H細胞的增殖對c-Src siRNA相比對c-Myc siRNA(對照)更為敏感,顯示c-Src網絡在這些細胞裏更加重要(圖4C)。結果顯示,c-Src(而非c-Myc)的消除導致Gucy1a3、Ticam1及Birc5(Survivin)(即c-Src網絡的所有組成部分)的表達被抑制,進一步證實了MA-10H細胞裏c-Src網絡的功能(圖4D)。總括而言,以上實驗證實,c-Src網絡在帶有Asp578His突變的LH信號轉導中擔起重要作用。
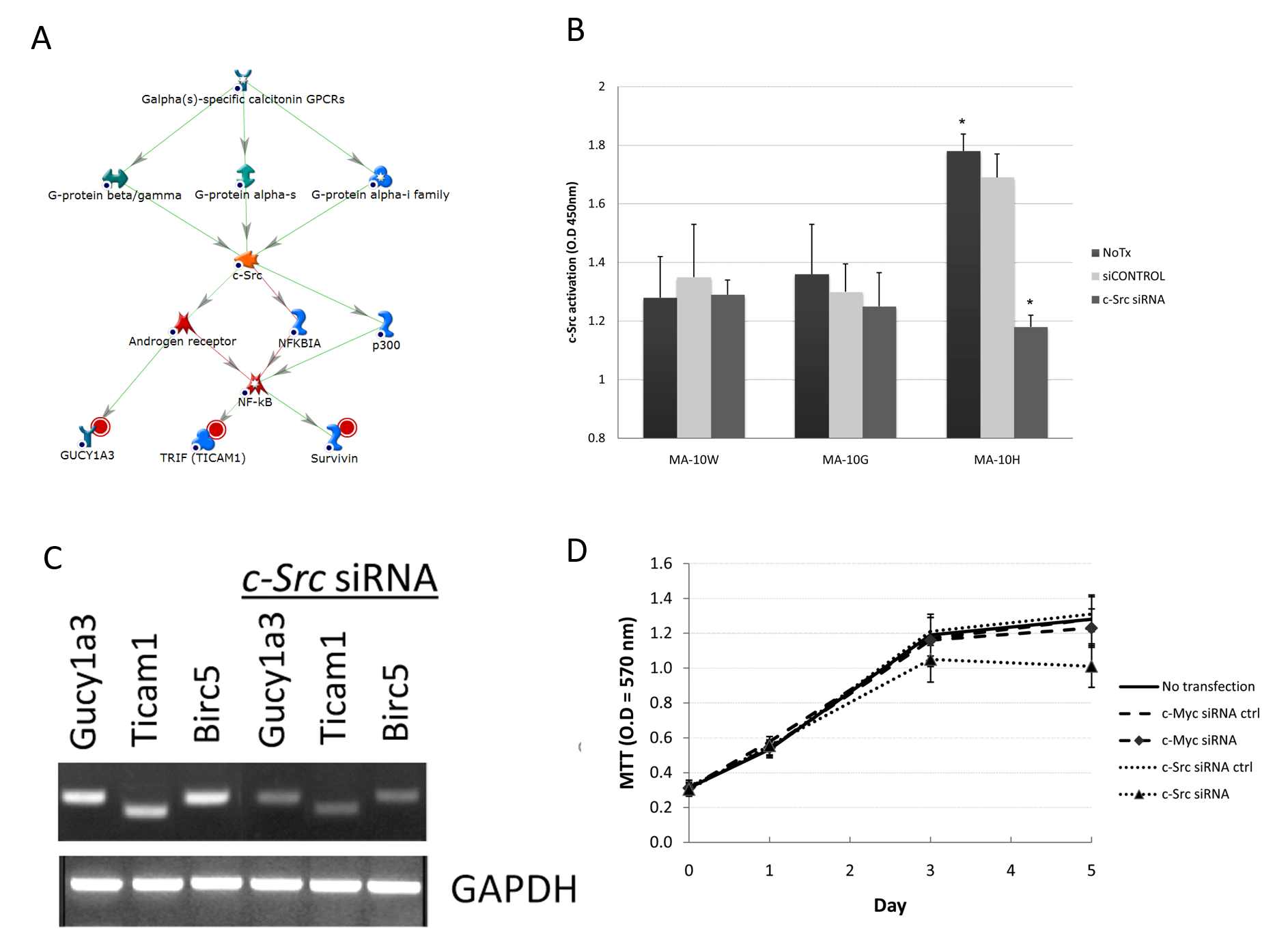
圖4A-D﹕﹕體細胞hLHR-Asp578His專屬c-Src信號網絡的確認。
A﹕MA-10H細胞內的c-Src網絡。紅色圓圈表代根節點。
B﹕c-Myc siRNA對MA-10G細胞內c-Myc活性的影響。如圖3B所述,我們透過暫時轉染100nM ON-TARGETplus siRNA(Dharmacon,Chicago,IL)來抑制c-Myc及c-Src。 結果發現比MA-10W及MA-10G更高的c-Src活性。即使c-Src siRNA轉染導致了MA-10G細胞內的c-Src活性降低,但MA-10H細胞仍對相同的處理更為敏感,其活性降低達33%以上(P < 0.05)。
C﹕c-Src對下游基因的調節。我們用c-Src siRNA轉染了細胞,然後進行半定量RT-PCR。我們在siRNA轉染之前及之後檢查了Gucy1a3、Ticam1及Birc5(Survivin)的表現。消除c-Src令Gucy1a3、Ticam1及Birc5(Survivin)的表現受到抑制,顯示MA-10H內c-Src網絡的所有組成部分均有運作。
D: 如圖3E之下所述,為c-Src對細胞增殖的特定作用的研究。結果顯示,MA-10H的增殖對c-Src siRNA比對c-Myc siRNA(對照)更加敏感,顯示c-Src途徑在MA-10H細胞內更為重要。
總結
本文的最初目的是闡述FMPP的病因。FMPP是由睾酮分泌異常所導致的男童性早熟病。基於對睾酮分泌的生理學認識,我們先將hLH水平與睾酮生物合成之間的關係切斷,並探索hLHR的活化突變、hLH介體,以及睾酮的合成機制。之後我們確立了一種可對所有FMPP病例作90%以上準確診斷的分子診斷方法。我們對FMPP患者的分子遺傳學作了廣泛研究,並用電腦為hLHR建立模型,再初步界定hLHR的跨膜螺旋信號轉導機制。然而,我們利用新建立的分子診斷方法,在一名早已確診的FMPP患者身上發現睾丸腫瘤,於是開展新一輪研究,探索FMPP、hLHR突變與睾丸腫瘤之間可能存在的關聯。該研究改變了對FMPP患者的治療方式,並推斷出有必要在患者進入青春期後頻繁召回,以便及早發現睾丸腫瘤。研究另外亦使我們發現hLHR的兩類活化突變,即生殖系突變及體細胞突變。hLHR的體細胞突變Asp578His不會導致FMPP,但會導致睾丸腫瘤。近期的表達譜分析及細胞模型研究發現到氨基酸578Asp裏有突變專屬網絡,能解釋特定突變的生殖系及體細胞性質。我們最初着手研究FMPP裏hLHR時並未設想到會出現以上曲折。研究過程的曲折離奇,峰迴路轉,或許能成為推動科學發現的某種動力。
鳴謝
本研究的部分支持來自中科院動物進化與遺傳前沿交叉卓越創新中心香港分部,VCDF CUHK No. 8601011。
參考
- Dufau ML. The luteinizing hormone receptor. In: The Leydig Cell (Payne AH, Hardy MP, Russell LD, Eds). Cache River Press, IL, pp. 333-350, 1996.
- Holland FJ. Gonadotropin-independent precocious puberty. Endocrinol Metab Clin North Amer. 20:191–210, 1991.
- Wu SM, Leschek EW, Rennert OM, Chan WY. Luteinizing hormone receptor mutations in disorders of sexual development and cancer. Frontiers Biosci. 5:D342-352, 2000.
- Nagasaki K, Katsumata N, Ogawa Y, Kikuchi T, Uchiyama M: Novel C617Y mutation in the 7th transmembrane segment of luteinizing hormone/choriogonadotropin receptor in a Japanese boy with peripheral precocious puberty. Endocr J. 57(12):1055-60, 2010.
- Wu SM, Leschek EW, Brain C, Chan WY. A novel activating mutation of the luteinizing hormone/chorionic gonadotropin receptor in a patient with FMPP - Effects of size versus charge. Mol Genet Metab. 66:68-73, 1999.
- Wu SM, Chan WY. Male pseudohermaphroditism due to inactivating luteinizing hormone receptor mutations. Arch Med Res. 30:495-500, 1999.
- Martin MM, Wu SM, Martin ALA, Rennert OM, Chan WY. Testicular seminoma in a patient with a constitutively activating mutation of the luteinizing hormone/chorionic gonadotropin receptor. Euro J Endocrinol. 139:101-106, 1998.
- Leschek EW, Chan WY, Diamond D, Laefer M, Jones J, Barnes KM, Cutler GB Jr. Nodular leydig cell hyperplasia in a boy with familial male-limited precocious puberty (FMPP). J Pediatr. 138:949-951, 2001.
- Liu G, Duranteau L, Monroe J, Doyle DA, Carel JC, Shenker A. Leydig-cell tumors caused by an activating mutation of the gene encoding the luteinizing hormone receptor. N Engl J Med. 341:1731-1735, 1999.
- Dang CV. Myc, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. Aug; 3(8): a014217, 2013.
- Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell. 6(3):209-214, 2004.
作者︰陳偉儀教授
理學士(中文大學)、PhD (FLA)
中文大學副校長
中文大學敬文書院院長
中文大學李嘉誠生物醫學講座教授
陳教授1974年在香港中文大學化學系一級榮譽畢業,於1977年在美國佛羅裡達大學取得生物化學哲學博士。1979年任美國俄克拉荷馬大學(University of Oklahoma)兒科學系及生物化學與分子生物學系助理教授,1983年升任副教授,並獲終身教職。1989年受聘美國喬治城大學(Georgetown University)任兒科學系及生物化學和分子與細胞生物學系教授並於翌年獲終身教職。2001年,借調到美國國立衛生研究院(NIH)國立兒童健康與人類發育研究所(NICHD),組建臨床基因組學研究室;於2006年1月獲委任為NICHD發育基因組學實驗室的主管及首席研究員。2009年應邀創立了香港中文大學生物醫學學院,擔任首任院長及生物醫學講座教授,並兼任中文大學組織工程及再生醫學研究所(iTERM)所長及中文大學─華大基因跨組學創新研究院院長。此外亦擔任中文大學與數所院校共建的聯合實驗室的主任,包括中國科學院廣州生物醫藥與健康研究院─中文大學再生生物學聯合實驗室,中文大學─中國科學院昆明動物研究所生物資源與疾病分子機理聯合實驗室,及中文大學─山東大學生殖遺傳聯合實驗室。2017年1月起出任中文大學敬文書院院長。
陳教授專注研究人類生殖及內分泌失衡病分子遺傳學,人類微量元素代謝病的生化遺傳學,生殖幹細胞及生殖系統腫瘤的功能性基因組及表觀遺傳學,非編碼RNA在多能幹細胞的分化中的作用與機理等。陳教授擁有七項發明專利,編輯和出版了五本學術專著,並出版了二十九篇課文及二百多篇學術論文。
陳教授擔任八本國際科學期刊的主編或副主編,及十六份國際科學期刊的編委。亦承擔過美國國立衛生研究院、英國維爾康基金健康研究委員會、愛爾蘭科學基金會、法國國立衛生和醫學研究所(INSERM)、澳門科學技術發展基金等機構的科研項目評審工作;也是香港大學教育資助委員會轄下的研究資助局評審委員會及香港食物及衛生局轄下的評審委員會委員。其他公職包括出任香港學術及職業資歷評審局項目評審專家等。陳教授亦參與香港及海外多個專業組織的工作,如曾任美洲華人遺傳學會主席及美國生殖學會發展委員會委員;現任美洲華人遺傳學會執行董事、香港科學會主席、邵逸夫獎理事會成員等。
陳教授是美國紐約華人醫學會榮譽會員及1997年喬治城大學傑出導師獎得主,並屢次被美國國立衛生研究院提名為傑出導師。其他榮譽包括1988年美國俄克拉荷馬醫學研究基金會傑出生物醫學研究獎、1989年俄克拉荷馬大學傑出貢獻獎、及2008年美洲華人遺傳學會總統獎。
2020年11月