简介
这个故事的主角是2007年邵逸夫生命科学与医学奖得主罗伯特‧尼科威教授的得奖研究,G蛋白偶联受体(G Protein-Coupled Receptors, GPCR)蛋白质家族的其中一个成员。尼科威教授后来亦因同一研究课题于2012年与布莱恩‧科比尔卡教授共同获得诺贝尔化学奖。GPCR是介导细胞及器官对药物和激素反应的主要受体系统。
故事源起于在一群性早熟男童里发现一种名为「家族性男性性早熟」(familial male-limited precocious puberty, FMPP)、具100%外显率的常染色体显性遗传病。患病的男童早至2至4岁就出现青春期特征,原因是其体内的男性激素睾酮达到成年男性的水平。而黄体生成激素(luteinising hormone, LH)量仅达青春期前的水平(LH是青春期时调节睾酮分泌量的激素)。由于异常情况是由睾酮过量分泌所导致,因此临床研究最初亦以睾酮中毒症(testotoxicosis)来描述此情况。由于已得知细胞表面受体——LH受体(LHR)会介导在睾酮分泌时LH的活动,因此研究人员检查了FMPP患者体内的受体,并发现LH受体出现活化突变。而研究亦意外发现FMPP患者患有另一种相对罕见的病症——睾丸肿瘤,以致亦发现了会引致肿瘤的LHR突变分支。这项发现改变了FMPP患者的传统临床治疗法。这是本次研究刚开始时,完全未预料到的结果。
黄体生成素/绒毛膜促性腺激素受体(Luteinising Hormone / Chorionic Gonadotropin Receptor, LHR)
LHR是GPCR家族的成员。这个蛋白质家族的成员一般有七个跨膜(transmembrane , TM)螺旋,这些螺旋会将与蛋白质胞外区结合的药物或激素的信号转导至细胞内的第二信使系统。激素与受体的结合会活化受体,引发一连串膜和细胞内活动。LHR有一个长N端细胞外区、七个跨膜α螺旋及一个短C端细胞内区1。人类LHR(hLHR)基因的编码区由10个内含子及11个外显子组成。前10个外显子为大部分细胞外区编码;外显子11则只为小部分N端细胞外区、全部7个TM区以及C端细胞内区编码。细胞外区乃负责LH的高亲和力结合。不同物种的TM区(包括TM螺旋及连接环)都高度一致,而TM区亦与其他糖激素受体高度一致。这个区以及胞质C端区对于转导引发第二信使系统活化的激素结合信号(腺苷酸环化酶级联反应)非常重要,会导致cAMP的合成增加,以及启动一连串膜和细胞内活动。而所引致的基因活化会增加睾酮分泌以及雄性性发育。由于同一受体还会与人类绒毛膜促性腺激素(human Chorionic Gonadotropin, hCG)结合,因此又称为黄体生成素/绒毛膜促性腺激素受体(Luteinising Hormone / Chorionic Gonadotropin Receptor, LH/hCGR,或简称hLHR)。大多数由LH信号异常引发的人类疾病都是hLHR突变的结果。
LHR的功能获得型活化突变–家族性男性性早熟(LHR—Familial Male-limited Precocious Puberty, FMPP)
人类的睾酮几乎全部由睾丸产生(超过95%)。正常生理条件之下,睾酮的主要来源是睾丸内的睾丸间质细胞。睾丸间质细胞表面的跨膜hLHR会介导LH的作用。由于hLHR在LH的作用下会触发睾酮的合成,因此我们先检查FMPP患者的hLHR。这个步骤找出了hLHR的单点活化突变,并确定了患者患上该病的病因。
FMPP是由活化突变所引致,而这些突变属功能获得型突变,并会引起男童的非性腺释素(luteinising-hormone releasing hormone, LHRH)依赖型同性早熟。hLHR的组成型活性会在无激素的情况下刺激胎儿时及青春期前的睾丸间质细胞,导致睾酮自行分泌。患病的男童最早会在3至4岁出现第二性特征、阴茎生长、双睾增大、阴毛生出,与真正的青春期无异。以往沿用的诊断法是量度睪酮水平。虽然患病男童的促性腺激素符合该年龄段的基础水平(即青春期前水平),但是他的睪酮却达到青春期至成年期的睾酮水平。该病之所以曾经名为睾酮中毒症,是因为当初在青春期前的患者身上测到达中毒水平的睾酮。如进行睾丸活体组织学检查,会发现睾丸间质细胞有生长2。hLHR的活化突变对女性患者无可见影响。治疗这种病可使用两种药物:螺内酯(spironolactone,抗雄激素药,能阻止睾酮起作用),以及睾内酯(testoleactone,抗芳香酶,能阻止睾酮形成)。进入青春期后,LH分泌会激增,触发睾酮的产生以及男性性发育,因此到青春期时会停用这两种药,以便天然产生的睾酮起作用。因此,当睾酮中毒患者进入自然青春期,一般均不会继续追纵。
FMPP的分子诊断
所有已知的FMPP突变都是在hLHR基因的外显子11(负责为TM区编码)上发现的单点错义突变。由于所有已知的hLHR活化突变都位于一个大外显子上,换言之可以利用聚合酶连锁反应(Polymerase chain reaction, PCR)进行DNA扩增然后测序,这种FMPP分子诊断方法会较为直接简单。在文献报导的120多名FMPP患者之中,超过50%是经由本人的实验室所确诊的。研究发现患者在15种氨基酸上出现18种不同的突变(图1A)3,4。一种氨基酸残基(Asp578)有3种不同的突变;另一种氨基酸残基(Asp564)则有2种不同的突变。约10%的突变是偶发突变(即新突变),而在FMPP家族中,有10%并无可见的hLHR异常状况。大多数FMPP突变出现在TM VI,因此TM VI是突变的热点。最常见的突变氨基酸是Asp-578。在所有活化突变之中,超过55%有影响该氨基酸。至于受家族及受新突变影响的患者,两者之间未见有临床表现上的差异。在非高加索族裔背景的患者,包括西班牙裔人、亚洲裔人、非洲裔美国人、非洲裔巴西人之中,稀有突变的发生率较高3。
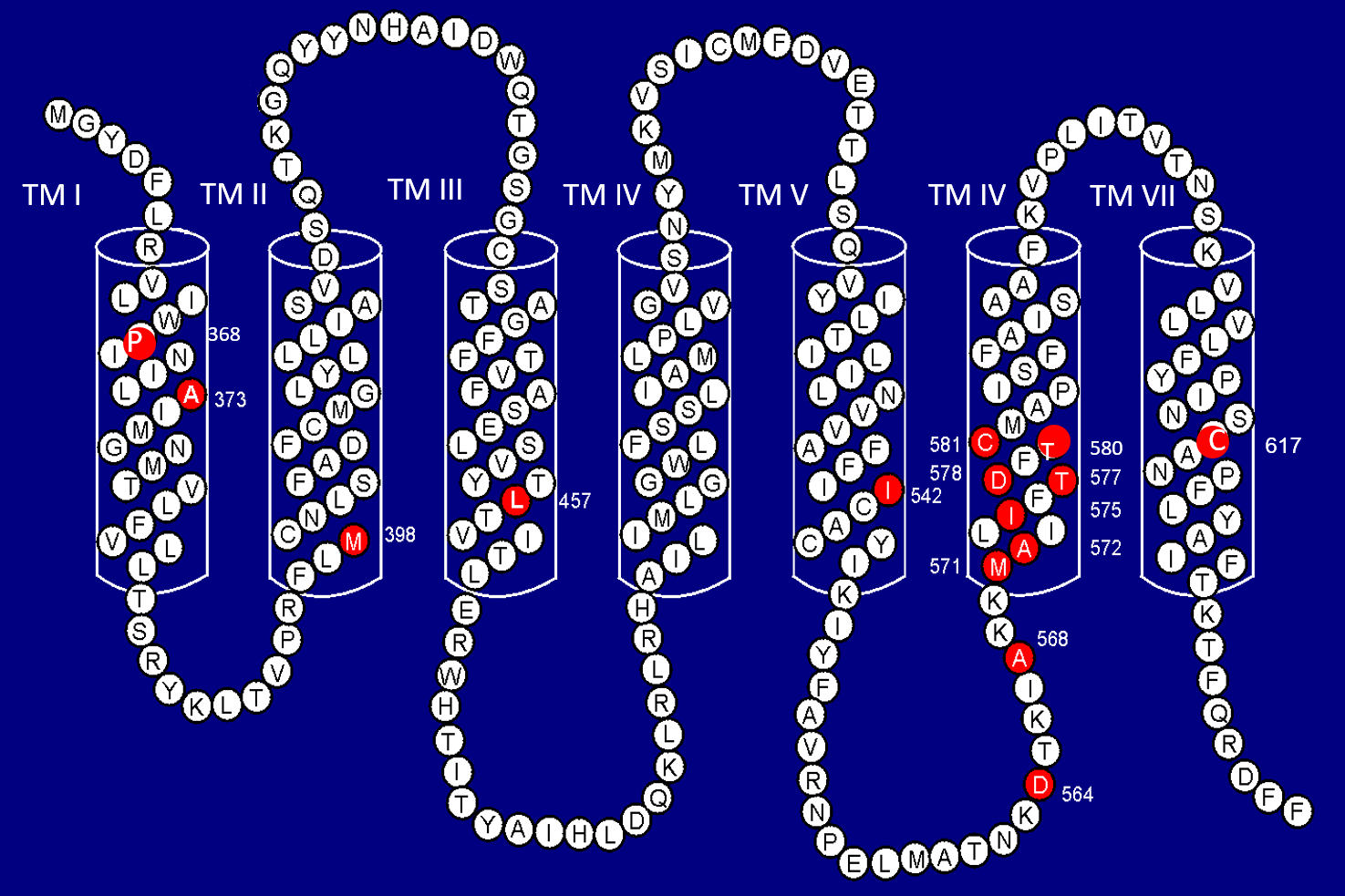
图1A﹕人类黄体生成素/绒毛膜促性腺激素受体(Human Luteinizing Hormone/Chorionic Gonadotropin Receptor, LHR)。
七个跨膜螺旋(TM I至TM VII) 红色实心圆圈代表在FMPP患者体内发现的突变氨基酸残基。氨基酸残基位置以数字表示。
由诊断至分子机制
人类黄体生成素受体(human luteinising hormone receptor, hLHR)以功能获得型突变活化,其机制大体上不明。我们调查hLHR的致病突变以及去活化突变后,得以利用结果进行hLHR的分子模型研究。大多数活化突变见于TM螺旋,尤其是hLHR的螺旋VI。进行分子模型研究,以及将TM VI与突变氨基酸并排作序列对比后,结果显示,受hLHR致病突变影响的氨基酸乃位于螺旋束的内部,指向由七个TM螺旋形成的导管之内(见图1B及表1)。对FMPP患者的Asp-578自然突变及定点诱变的研究显示,由Asp-578突变引起的hLHR活化,其活化程度大体上取决于更代氨基酸的侧链大小,而非其电荷5。因此,这些氨基酸的突变将改变螺旋间相互作用以及导致活化的受体的构形, 而这种改变可能实际涉及由螺旋形成的导管的张开。
因此得出结论﹕Asp-578在hLHR的信号转导中担当位置恰当的氢键受体,发挥重要作用。我们对位于螺旋之外的氨基酸残基进行了定点诱变研究,结果显示其他类型的相互作用(例如静电及空间相互作用)对于受体的活化亦可能有重要作用5。
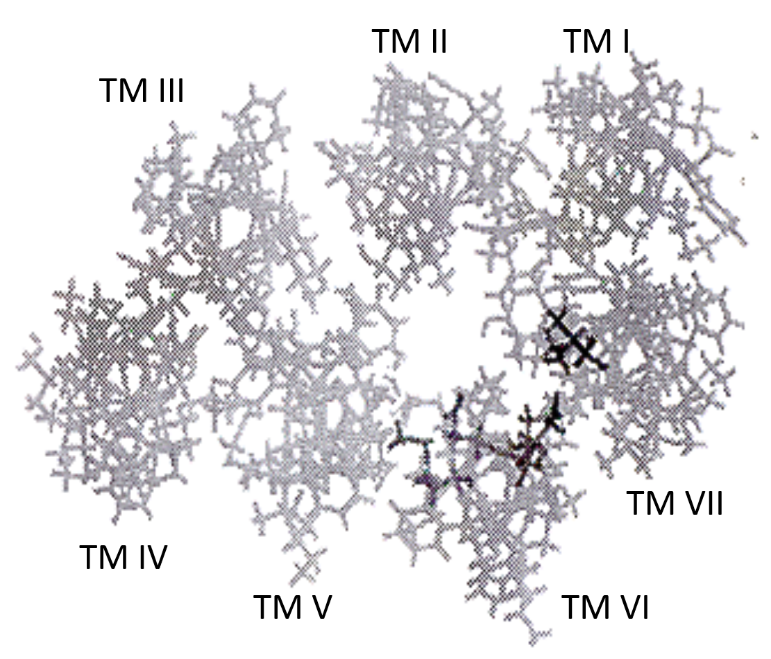
图1B﹕人类黄体生成素/绒毛膜促性腺激素受体(Human Luteinizing Hormone/Chorionic Gonadotropin Receptor, LHR)。
跨膜螺旋电脑模型鸟瞰图。较深色符号代表突变氨基酸残基。
| 正常 AA | 突变 | 位置 # | 螺旋转角 # (3.6 AA/转角) |
疾症 |
|---|---|---|---|---|
| Met-571 | Ile | 1 | 0 | FMPP |
| Ala-572 | Val | 2 | 0 | FMPP |
| Ile -575 | Leu | 5 | 1 | FMPP |
| Thr-577 | Ile | 7 | 2 | FMPP |
| Asp-578 | Gly/Tyr/Glu | 8 | 3 | FMPP |
| Cys-581 | Arg | 11 | 4 | FMPP |
| Ile-585 | No known | 14 | 5 | - |
| Ala-589 | No known | 19 | 6 | - |
| Ala-593 | Pro | 23 | 7 | LCH |
表1﹕TM VI内各种突变氨基酸残基(AA)的并列比较。
并无检测到氨基酸残基Ile-585及Ala-589的突变。
另一个极端——功能丧失型失活突变——睾丸间质细胞发育不良(Leydig Cell Hypolasia, LCH)
另一方面,类似的错义突变亦可使受体失活,导致男性性发育低于正常水平。失活突变属功能丧失型突变,会导致受体无法与激素结合或已结合的激素无法活化受体。这类突变会影响性发育,特别是对男性。当男性体内发生纯合子或复合杂合子失活突变时,hLHR活性的降低或废止会导致对LH刺激产生抗性,令睾丸间质细胞无法分化,于是出现FMPP的相反——睾丸间质细胞发育不良(LCH)或睾丸间质细胞不发育(Leydig Cell Agenesis),是假性雌雄同体(pseudohermaphroditism)之一种。LCH的临床表现并不一致。一边极端是遗传上是男性但长有外观正常的女性外生殖器,他们最后往往会接受变女性手术。另一边极端的个案则是患有高促性腺素性功能减退症(hypergonadotropic hypogonadism)、小阴茎以及男性外生殖器发育不良。而两者之间的个案则是程度各异的外生殖器男性化。由于任何突变都有可能使已编码的蛋白质失活,因此LCH的分子遗传学表现比FMPP更为不一。在LCH患者中发现的hLHR失活突变包括错义突变、无义突变、插入、缺失。另外在若干LCH个案中并未发现有hLHR突变,表示失活突变的样式繁多,或者LCH是由hLHR以及其他基因的突变所引起。我们所观察到的遗传突变有多种形态表现,同样,各种突变对hLHR作用的影响亦各有不同。视乎受体蛋白中突变位置的不同,突变可导致激素的结合减少、表面表现减少、细胞内转运异常,或偶联效率降低,所有情况都会导致激素结合所触发的信号转导减少或缺失6。
新的波折——癌症中的生殖系突变及体细胞活化突变
一般而言,当对某疾病作分子遗传学研究时,能设计出诊断方法以及解构出疾病的机制已可以称得上是达到研究的目标了。然而,FMPP的研究却出现另一番波折。有一位患者在27个月大时在乔治城大学医学中心根据内分泌评估被诊断患有FMPP,后来到35岁时获诊断出有睾丸精原细胞瘤。为确认该患者的FMPP诊断,他接受了对hLHR基因的分子分析,并发现体内有活化突变Asp578Gly。这是首宗确诊FMPP患者患上睾丸肿瘤的报告7。在此报告之后,又发现另一名FMPP患者出现活化突变Asp564Gly,而且长有两个无症状睾丸间质细胞瘤8。由于突变是在循环血细胞基因组DNA中检测得到,因此两个个案的hLHR突变都属生殖系。在第一位FMPP患者被诊断出有睾丸精原细胞瘤一年后,Liu等人9报告有三名患者在睾丸间质细胞腺瘤组织内的hLHR基因中出现活化突变Asp578His。该项突变并未在相邻的正常组织或血细胞之中出现。因此,尽管未知高水平的睾酮产生会否导致肿瘤形成,但是具有组成性活性的hLHR或会使人易患睾丸肿瘤。医学界以往会在FMPP患者进入自然青春期后送其出院,这项发现改变了传统的做法,并提出有必要在患者进入青春期之后继续追踪,检查他们是否出现早期睾丸肿瘤。
hLHR的突变偏向信号途径
研究hLHR突变影响的细胞模型
我们在FMPP患者中从未检测到Asp578His突变;这种突变乃体细胞突变,不会经生殖细胞传递至下一代。Asp578His的致癌机制尚未得知。以上观察结果为hLHR活化突变的生物效应研究开闢了新的方向。
为探究活化突变的遗传效应,我们建立了一套细胞模型。我们利用老鼠睾丸间质细胞系MA-10细胞(这细胞还保有睾丸间质细胞部分生成类固醇及对LH的刺激有反应的性质)来比较Asp578Gly与Asp578His突变对睾丸间质细胞的影响。我们使用了野生型hLHR(hLHR-WT)的重组pcDNA3质体、带有Asp578Gly(hLHR-D578G)或Asp578H(hLHR-D578H)的hLHR来转染MA-10细胞。我们使用了老鼠全基因组芯片来检查三种细胞的表达谱,即MA-10W(表达hLHR-WT的MA-10细胞)、MA-10G(表达hLHR-D578G的MA-10细胞)及MA-10H(表达hLHR-D578H的MA-10细胞)。如图2A所示,我们用无引导阶层式分群法分析了野生型及突变型转染子(每个类别3个复制)所表达的基因。分群法将表达谱明显分成三组,与所表现的hLHR基因型一致。MA-10G及MA-10H与对照的MA-10W细胞迥然不同。相比MA-10W,MA-10G及-10H有132个调升的基因。其中大多数是癌基因以及与发育(特别是神经元发育)有关的基因。我们对MA-10W与两个突变细胞(MA-10G及MA-10H)之间表达差异的基因进行了本体分析,发现两个突变体有共同的七个生物学类别(表2)。然而,两类在MA-10G细胞发现的基因(即与发育成熟及繁殖有关的基因)在MA-10H突变体中并无出现, 因此确证了Asp578His突变的体细胞特性(该特性在FMPP患者中尚未发现)。
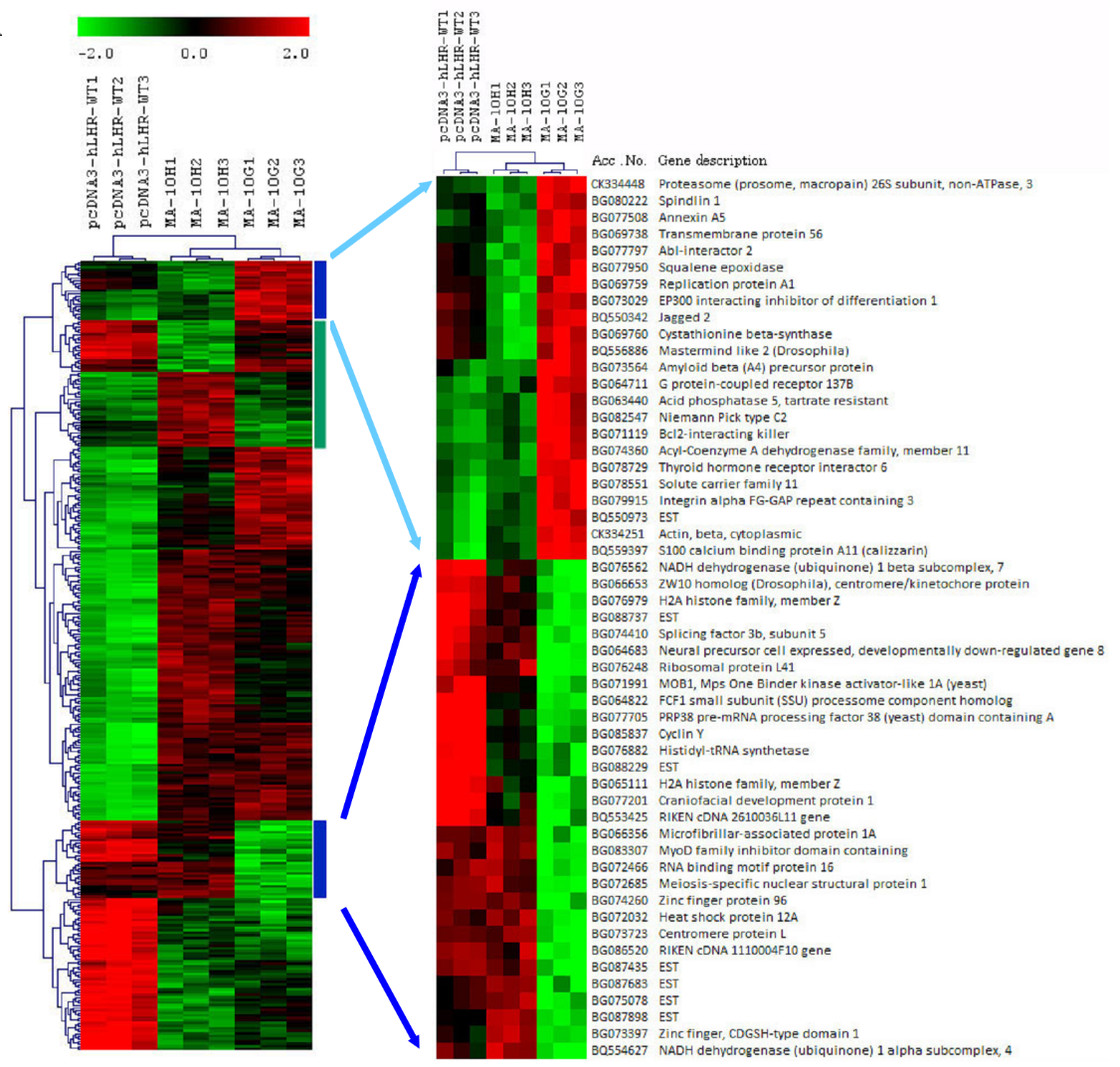
图2A﹕MA-10转染子的阶层式分群法及突变专属网路的鑑认。
MA-10W(pcDNA3-hLHR-WT,复制1至3)、MA-10H(hLHR-D578H,复制1至3)及MA-10G(hLHR-D578G,复制1至3)里的已表达基因阶层分群 。蓝色直线代表带有与MA-10G细胞相关的表达特征的基因群;绿色直线代表带有与MA-10H细胞相关的表达特征的基因群。为显示基因名称,特别放大蓝线簇群。
| hLHR-Asp578Gly (MA-10G) | hLHR-Asp578His (MA-10H) |
|---|---|
| 细胞代谢过程 | 细胞代谢过程 |
| 发育 | 发育 |
| 生长 | 生长 |
| 生物之间的相互作用 | 生物之间的相互作用 |
| 生理过程 | 生理过程 |
| 生物过程调节 | 生物过程调节 |
| 对刺激的反应 | 对刺激的反应 |
| 发育成熟 | - |
| 繁殖 | - |
表2﹕MA-10G及MA-10H细胞根据基因本体分析表达谱的主要生物学类别分析
当比较MA-10G与MA-10H细胞的表达谱时,会发现在两个突变细胞之间表现相异的两个特征基因群(图2A,蓝色条及绿色条),表示该两个突变hLHR的基因组效应并不相同。由此可见,用两种不同的氨基酸替换同一种氨基酸,会对相关基因的表达产生不同影响,是为hLHR里有突变偏向基因表达的第一项证据。
我们试图透过重新生成由基因群代表的细胞网路来分辨与MA-10G及MA-10H突变体有关的网路。为了在生物网路的环境里将基因表达资料视觉化,我们利用了由多个来源集合而成的GeneGo MetaCore资料库分析基因群,以便查询及检索生物学感兴趣的基因与蛋白质之间关系,并在集成网路上获得使生物学假设成立的拓扑不变量(topological property)。上述分析产生了分别与MA-10G细胞及MA-10H细胞相关的9及12个网路。为了减低两个突变体共佔融合网路的可能性,我们消除了两个网路共有的所有基因元素。该过程分别产生了两个及三个MA-10G细胞及MA-10H细胞专属的网路(图2B)(表3)。突变专属的网路只涉及调升的基因。
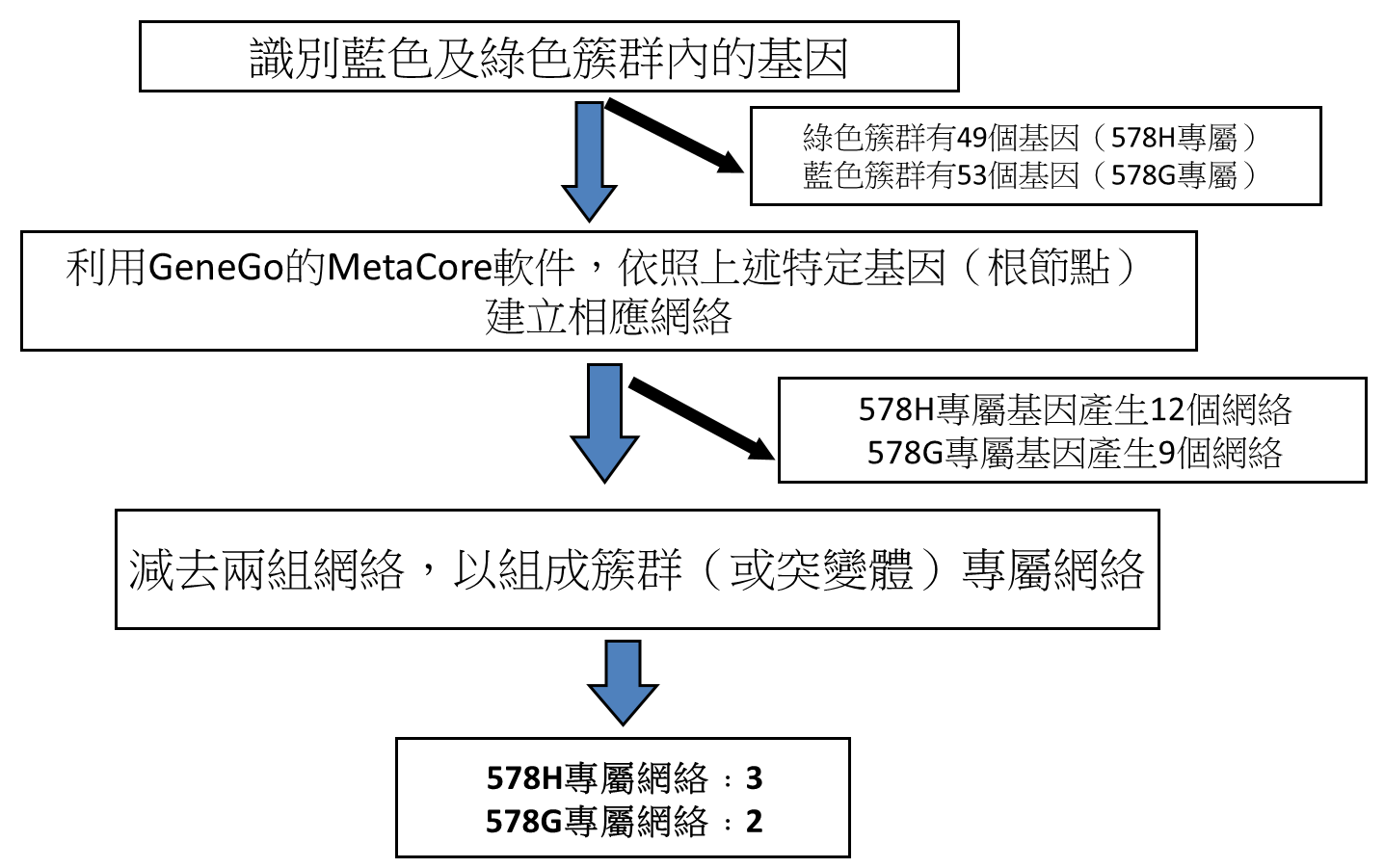
图2B﹕MA-10转染子的阶层式分群法及突变专属网路的鑑认。
GeneGo的MetaCore分析,用作识别突变专属网路。
| 突变基因类型 | 网路 | (调节子) 根节点 |
|---|---|---|
| Asp578Gly | 1 | (c-Myc) APP、Jagged2、CFDP1、H2AFZ、CBS |
| 2 | (PKC-alpha) APP-C59,膜联蛋白 | |
| Asp578His | 1 | (c-Src)GUCY1A3,TRIF(TICAM1),Survivin |
| 2 | (nAChR)AARS,鸟苷酸环化酶,钙调蛋白 | |
| 3 | (ESR1) MIF,转移蛋白,C8orf40,肾上腺髓质素 |
表3﹕发现突变专属网路。
生殖细胞hLHR-Asp578Gly专属信号网路的确认
为了确认MA-10G或MA-10H细胞内有网路存在,我们使用了siRNA来检查关键成员(调节器)对网路里其他根节点以及对相应突变细胞之增殖的消除效果。举例而言,我们的研究结果显示,在两个MA-10G专属网路之一里,c-Myc(一种在控制细胞增殖、分化、凋亡及维持细胞内平衡中发挥重要作用的转录因数)可以利用与发育相关的多种功能(例如β类淀粉蛋白(A4)前体蛋白(App)、颅面发育蛋白1(Cfdp1)及胱硫醚β合酶(Cbs)来调节下游基因的表达,以及利用H2A组蛋白家族(H2afz)来调节对DNA结构完整度或基因活化10的维持(图3A)。相比MA-10W(56%)及MA-10H(64%)细胞,MA-10G表现出更高的基础c-Myc活性,并且对c-Myc siRNA抑制更敏感,因为其活性降低达40%(p < 0.05)(图3B)。如表达谱分析所显示,c-Myc是由Notch1调节;Notch1则是Jagged2的一个下游标靶;Jagged2则是一个被hLHR-D578G上调的基因。蛋白转渍法(Western blotting)的结果证实了Jagged2对Notch1的调节;当中可见,用Jagged2 siRNA处理后,MA-10G细胞里的Notch1裂解水平降低,Notch1受到抑制;但在MA-10H细胞里,由于正常情况下Notch1蛋白量很少,因此并未明显受抑制(图3C)。实验证实了c-Myc网路在MA-10G细胞里的活性,因为 c-Myc的消除可导致c-Myc网路的所有组成部分(即App、Cfdp1、H2afz及Cbs)的表达被抑制(图3D)。图3E亦显示,MA-10G细胞的增殖对c-Myc siRNA比对c-Src siRNA(对照)更为敏感,即是c-Myc网路在该等细胞中更为重要。总括而言,以上实验显示,hLHR-D578G的专属网路之一是透过生殖系活化突变来上调Jagged2,此乃Notch1 – c-Myc调节作用的开关,并导致细胞增殖增加。
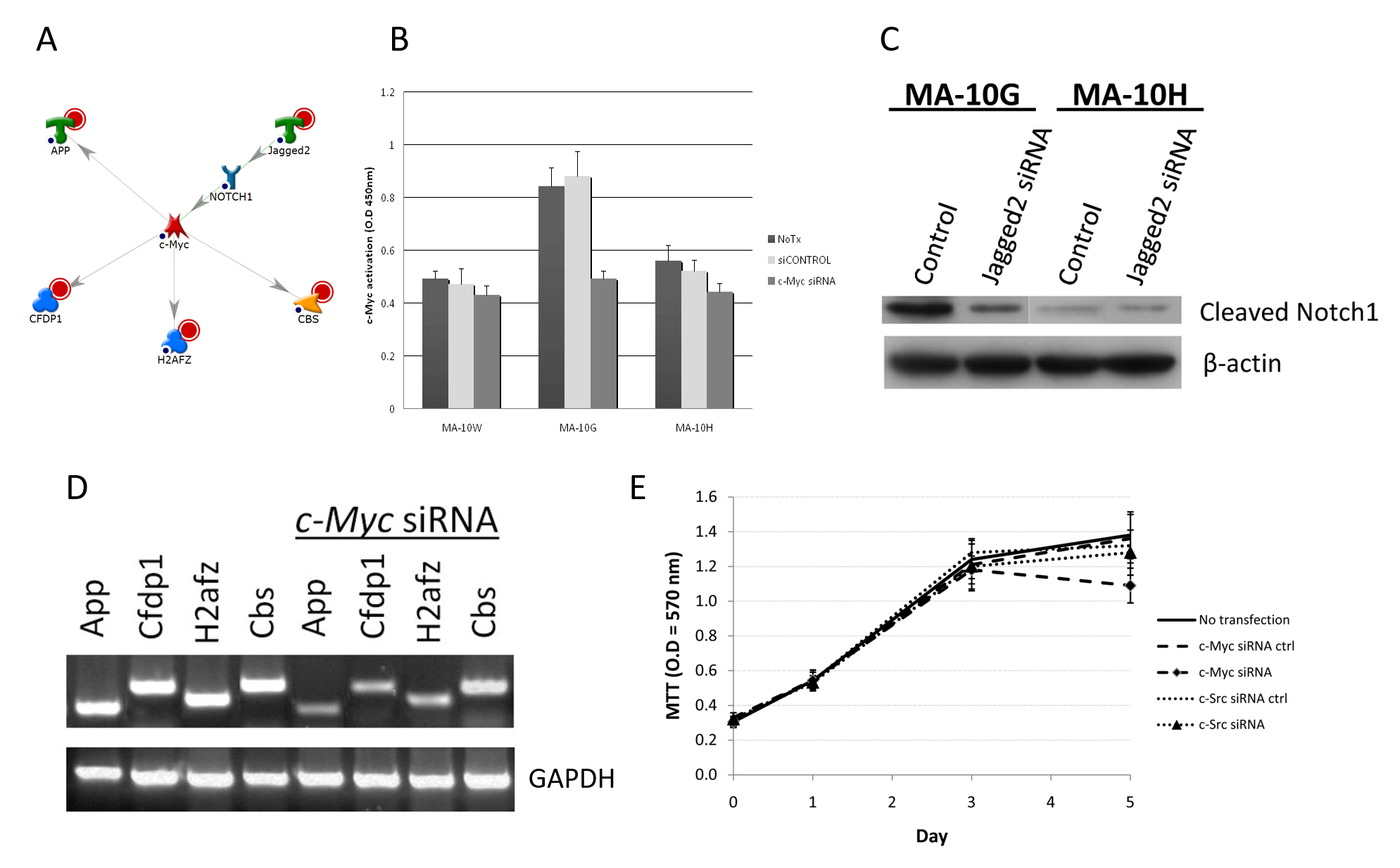
图3A-E﹕生殖系hLHR-Asp578Gly专属c-Myc信号网路的确认。
A:MA-10G细胞内c-Myc网路。红色圆圈代表根节点。
B:c-Myc siRNA对MA-10G细胞内c-Myc活性的影响。根据厂商指引,短暂转染100nM ON-TARGETplus siRNA(Dharmacon,Chicago,IL)24小时来抑制c-Myc及c-Src。为了确认个别siRNA在消除时的特异性,每次转染实验都设有ON-TARGETplus siCONTROL非标靶对照(Dharmacon,Chicago,IL)。使用TransAM c-Myc及FACE c-Src试剂盒(Active Motif,Carlsbad,CA)为活化的c-Myc及c-Src信号蛋白作定量。利用分光光度计将光密度定为450 nM,透过比色反应量度活化信号。MA-10G表现出较高的基础c-Myc活性,并对c-Myc的抑制作用更为敏感,因为其活性降低达40%(p < 0.05)。
C:以Notch1为介体之Jagged2对c-Myc调节的确认。利用GO及网路分析检查MA-10G细胞内Jagged2调节c-Myc之时Notch1的参与。配体结合后,Notch受体被裂解,并释放出一种细胞内亚基;该亚基迁移至细胞核,并在细胞核内担当转录辅激活蛋白的作用。因此,我们以蛋白转渍法检查了MA-10G及MA-10H细胞内有或无Jagged2抑制作用时的Notch1裂解量。在MA-10G细胞内观察到裂解的Notch1明显减少(MA-10H则无)。
D:c-Myc对下游基因的调节。我们用c-Myc siRNA转染细胞,然后进行半定量RT-PCR。我们在siRNA转染前后均检查了App、Cfdp1、H2afz及Cbs的表现。c-Myc的消除导致pp、Cfdp1、H2afz及Cbs的表达被抑制,可见MA-10G内c-Myc网路的所有组成部分均有运行。
E:为了研究野生型或突变型hLHR对c-Myc的细胞增殖的特定作用,我们利用c-Myc siRNA转染进行了五日的3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四唑溴化物(MTT)化验。MA-10G的增殖对c-Myc siRNA的敏感度比对c-Src siRNA(对照)高,显示c-Myc途径在MA-10G细胞内更为重要。
体细胞hLHR-Asp578His专属信号网路的确认
在三个MA-10H专属网路之一里,H专属簇基因鸟苷酸环化酶1 alpha 3(Gucy1a3)、类铎受体衔接子分子(Ticam1)及生存素(Birc5)是透过c-Src的控制而受调节(c-Src是一种已知的G蛋白受体标靶以及转录因数NF-κB的调节子)(图4A)。C-Src(原癌基因酪氨酸蛋白激酶Src)是一种脂蛋白信号分子(经荳蔻酰化),透过多种底物的磷酸化参与细胞间相互作用、细胞迁移及增殖11。至于MA-10H细胞,研究发现其c-Src活性比MA-10W及MA-10G高,分别高26%及27%(图4B)。尽管利用c-Src siRNA作转染导致了MA-10G细胞里的c-Src活性降低,但MA-10H细胞对相同的处理更为敏感,活性降低达33%(P < 0.05)。这项结果与先前对c-Myc的研究共同证实了c-Myc在MA-10G以及c-Src在MA-10H细胞中的主要调节作用(图4B)。MA-10H细胞的增殖对c-Src siRNA相比对c-Myc siRNA(对照)更为敏感,显示c-Src网路在这些细胞里更加重要(图4C)。结果显示,c-Src(而非c-Myc)的消除导致Gucy1a3、Ticam1及Birc5(Survivin)(即c-Src网路的所有组成部分)的表达被抑制,进一步证实了MA-10H细胞里c-Src网路的功能(图4D)。总括而言,以上实验证实,c-Src网路在带有Asp578His突变的LH信号转导中担起重要作用。
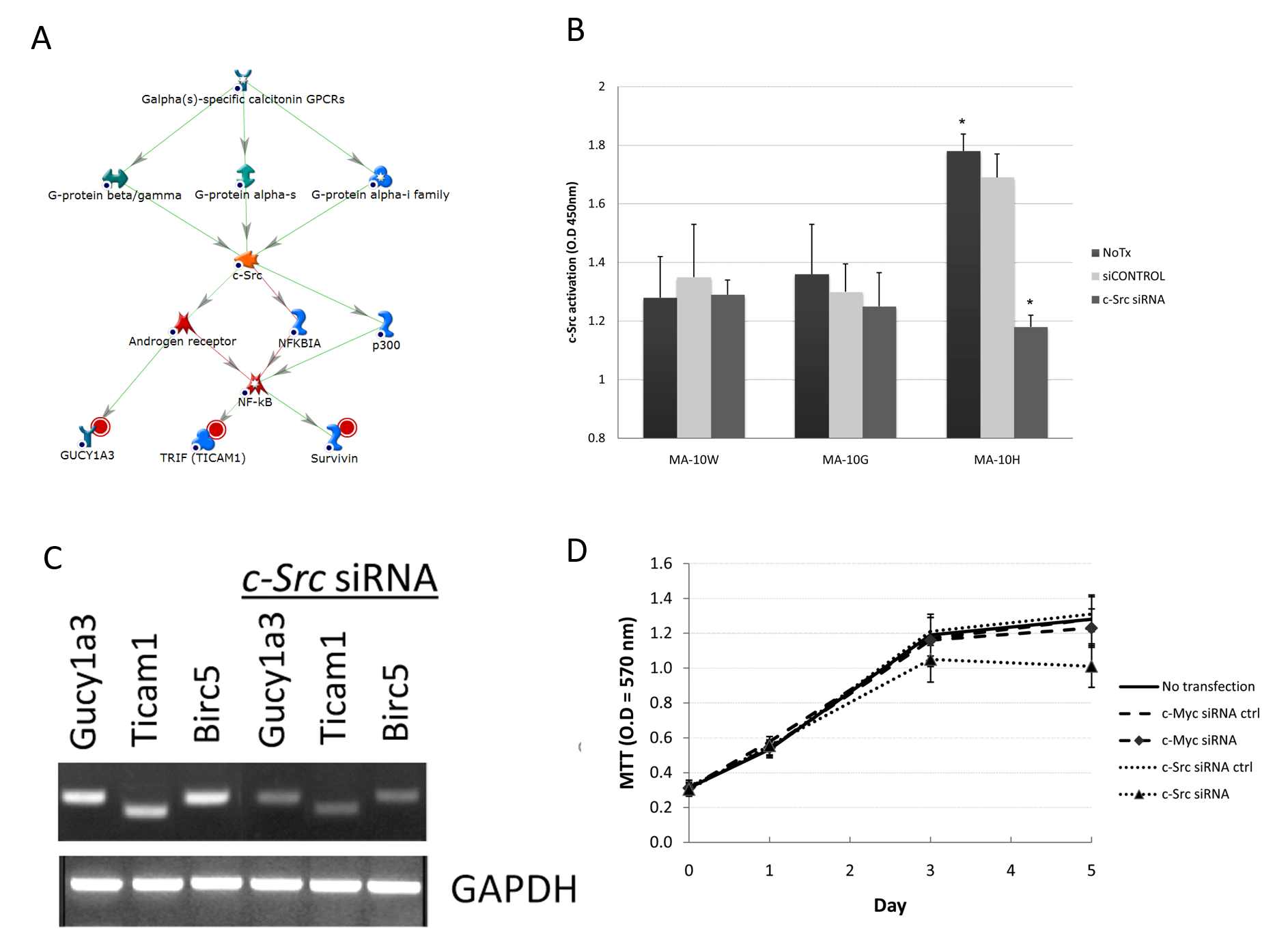
图4A-D﹕﹕体细胞hLHR-Asp578His专属c-Src信号网路的确认。
A﹕MA-10H细胞内的c-Src网路。红色圆圈表代根节点。
B﹕c-Myc siRNA对MA-10G细胞内c-Myc活性的影响。如图3B所述,我们透过暂时转染100nM ON-TARGETplus siRNA(Dharmacon,Chicago,IL)来抑制c-Myc及c-Src。 结果发现比MA-10W及MA-10G更高的c-Src活性。即使c-Src siRNA转染导致了MA-10G细胞内的c-Src活性降低,但MA-10H细胞仍对相同的处理更为敏感,其活性降低达33%以上(P < 0.05)。
C﹕c-Src对下游基因的调节。我们用c-Src siRNA转染了细胞,然后进行半定量RT-PCR。我们在siRNA转染之前及之后检查了Gucy1a3、Ticam1及Birc5(Survivin)的表现。消除c-Src令Gucy1a3、Ticam1及Birc5(Survivin)的表现受到抑制,显示MA-10H内c-Src网路的所有组成部分均有运作。
D: 如图3E之下所述,为c-Src对细胞增殖的特定作用的研究。结果显示,MA-10H的增殖对c-Src siRNA比对c-Myc siRNA(对照)更加敏感,显示c-Src途径在MA-10H细胞内更为重要。
总结
本文的最初目的是阐述FMPP的病因。FMPP是由睾酮分泌异常所导致的男童性早熟病。基于对睾酮分泌的生理学认识,我们先将hLH水平与睾酮生物合成之间的关系切断,并探索hLHR的活化突变、hLH介体,以及睾酮的合成机制。之后我们确立了一种可对所有FMPP病例作90%以上准确诊断的分子诊断方法。我们对FMPP患者的分子遗传学作了广泛研究,并用电脑为hLHR建立模型,再初步界定hLHR的跨膜螺旋信号转导机制。然而,我们利用新建立的分子诊断方法,在一名早已确诊的FMPP患者身上发现睾丸肿瘤,于是开展新一轮研究,探索FMPP、hLHR突变与睾丸肿瘤之间可能存在的关联。该研究改变了对FMPP患者的治疗方式,并推断出有必要在患者进入青春期后频繁召回,以便及早发现睾丸肿瘤。研究另外亦使我们发现hLHR的两类活化突变,即生殖系突变及体细胞突变。hLHR的体细胞突变Asp578His不会导致FMPP,但会导致睾丸肿瘤。近期的表达谱分析及细胞模型研究发现到氨基酸578Asp里有突变专属网路,能解释特定突变的生殖系及体细胞性质。我们最初着手研究FMPP里hLHR时并未设想到会出现以上曲折。研究过程的曲折离奇,峰回路转,或许能成为推动科学发现的某种动力。
鸣谢
本研究的部分支持来自中科院动物进化与遗传前沿交叉卓越创新中心香港分部,VCDF CUHK No. 8601011。
参考
- Dufau ML. The luteinizing hormone receptor. In: The Leydig Cell (Payne AH, Hardy MP, Russell LD, Eds). Cache River Press, IL, pp. 333-350, 1996.
- Holland FJ. Gonadotropin-independent precocious puberty. Endocrinol Metab Clin North Amer. 20:191–210, 1991.
- Wu SM, Leschek EW, Rennert OM, Chan WY. Luteinizing hormone receptor mutations in disorders of sexual development and cancer. Frontiers Biosci. 5:D342-352, 2000.
- Nagasaki K, Katsumata N, Ogawa Y, Kikuchi T, Uchiyama M: Novel C617Y mutation in the 7th transmembrane segment of luteinizing hormone/choriogonadotropin receptor in a Japanese boy with peripheral precocious puberty. Endocr J. 57(12):1055-60, 2010.
- Wu SM, Leschek EW, Brain C, Chan WY. A novel activating mutation of the luteinizing hormone/chorionic gonadotropin receptor in a patient with FMPP - Effects of size versus charge. Mol Genet Metab. 66:68-73, 1999.
- Wu SM, Chan WY. Male pseudohermaphroditism due to inactivating luteinizing hormone receptor mutations. Arch Med Res. 30:495-500, 1999.
- Martin MM, Wu SM, Martin ALA, Rennert OM, Chan WY. Testicular seminoma in a patient with a constitutively activating mutation of the luteinizing hormone/chorionic gonadotropin receptor. Euro J Endocrinol. 139:101-106, 1998.
- Leschek EW, Chan WY, Diamond D, Laefer M, Jones J, Barnes KM, Cutler GB Jr. Nodular leydig cell hyperplasia in a boy with familial male-limited precocious puberty (FMPP). J Pediatr. 138:949-951, 2001.
- Liu G, Duranteau L, Monroe J, Doyle DA, Carel JC, Shenker A. Leydig-cell tumors caused by an activating mutation of the gene encoding the luteinizing hormone receptor. N Engl J Med. 341:1731-1735, 1999.
- Dang CV. Myc, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. Aug; 3(8): a014217, 2013.
- Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell. 6(3):209-214, 2004.
作者︰陈伟仪教授
理学士(中文大学)、PhD (FLA)
中文大学副校长
中文大学敬文书院院长
中文大学李嘉诚生物医学讲座教授
陈教授1974年在香港中文大学化学系一级荣誉毕业,于1977年在美国佛罗里达大学取得生物化学哲学博士。1979年任美国俄克拉荷马大学(University of Oklahoma)儿科学系及生物化学与分子生物学系助理教授,1983年升任副教授,并获终身教职。1989年受聘美国乔治城大学(Georgetown University)任儿科学系及生物化学和分子与细胞生物学系教授并于翌年获终身教职。2001年,借调到美国国立卫生研究院(NIH)国立儿童健康与人类发育研究所(NICHD),组建临床基因组学研究室;于2006年1月获委任为NICHD发育基因组学实验室的主管及首席研究员。2009年应邀创立了香港中文大学生物医学学院,担任首任院长及生物医学讲座教授,并兼任中文大学组织工程及再生医学研究所(iTERM)所长及中文大学─华大基因跨组学创新研究院院长。此外亦担任中文大学与数所院校共建的联合实验室的主任,包括中国科学院广州生物医药与健康研究院─中文大学再生生物学联合实验室,中文大学─中国科学院昆明动物研究所生物资源与疾病分子机理联合实验室,及中文大学─山东大学生殖遗传联合实验室。2017年1月起出任中文大学敬文书院院长。
陈教授专注研究人类生殖及内分泌失衡病分子遗传学,人类微量元素代谢病的生化遗传学,生殖干细胞及生殖系统肿瘤的功能性基因组及表观遗传学,非编码RNA在多能干细胞的分化中的作用与机理等。陈教授拥有七项发明专利,编辑和出版了五本学术专着,并出版了二十九篇课文及二百多篇学术论文。
陈教授担任八本国际科学期刊的主编或副主编,及十六份国际科学期刊的编委。亦承担过美国国立卫生研究院、英国维尔康基金健康研究委员会、爱尔兰科学基金会、法国国立卫生和医学研究所(INSERM)、澳门科学技术发展基金等机构的科研项目评审工作;也是香港大学教育资助委员会辖下的研究资助局评审委员会及香港食物及卫生局辖下的评审委员会委员。其他公职包括出任香港学术及职业资历评审局项目评审专家等。陈教授亦参与香港及海外多个专业组织的工作,如曾任美洲华人遗传学会主席及美国生殖学会发展委员会委员;现任美洲华人遗传学会执行董事、香港科学会主席、邵逸夫奖理事会成员等。
陈教授是美国纽约华人医学会荣誉会员及1997年乔治城大学杰出导师奖得主,并屡次被美国国立卫生研究院提名为杰出导师。其他荣誉包括1988年美国俄克拉荷马医学研究基金会杰出生物医学研究奖、1989年俄克拉荷马大学杰出贡献奖、及2008年美洲华人遗传学会总统奖。
2020年11月