Introduction
The central character of the story is a member of a family of proteins for which Professor Robert J Lefkowitz was awarded the Shaw Prize in Life Science & Medicine in 2007, and then in 2012, shared with Professor Brian K Kobilka the Nobel Prize in Chemistry. The family is the G Protein-Coupled Receptors (GPCR), the major receptor system that mediates the response of cells and organs to drugs and hormones.
In a group of boys with precocious puberty, an autosomal dominant disorder with 100% penetrance, called Familial Male-limited Precocious Puberty (FMPP) is identified. Early onset of pubertal changes in these boys occur as young as 2-4 years of age triggered by the production of adult level of the male sex hormone, testosterone. On the other hand, prepubertal level of luteinising hormone (LH), the hormone responsible for regulating the production of testosterone at puberty, is observed. Since the abnormality is due to over production of testosterone, the disorder was also called Testotoxicosis when first described clinically. Knowledge of the role of a cell surface receptor, the LH receptor (LHR) in mediating the action of LH in the production of testosterone led to the examination of the receptor in FMPP patients and the subsequent discovery of activating mutations of the receptor. Accidental identification of FMPP patients developing another relatively rare pathological condition, testicular tumor, led to the discovery of a subset of LHR mutations that are tumorigenic. This discovery changes the traditional clinical management of FMPP patients. The final outcome of the story is totally unexpected when the study first started.
Luteinising Hormone / Chorionic Gonadotropin Receptor (LHR)
The LHR is a member of the GPCR family. This family of proteins typically have 7 transmembrane (TM) helices which transduce the signal of drug or hormone binding to the extracellular domain of the protein to the second messenger system inside the cell. Binding of the hormone to the receptor activates the receptor leading to a sequence of membrane and intracellular events. LHR has a long N-terminal extracellular domain, 7 membrane-spanning α helices and a short C-terminal intracellular domain1. The coding region of the human LHR (hLHR) gene consists of 10 introns and 11 exons. The first 10 exons encode the majority of the extracellular domain, while exon 11 codes for only a small portion of the N-terminal extracellular domain, all 7 TM domains, and the C-terminal intracellular domain. The extracellular domain is responsible for high affinity binding of LH. The TM domain, including the TM helices and the connecting loops, is highly homologous between species and with the other glycohormone receptors. This domain and the cytoplasmic C-terminal domain are important for transducing the signal of hormone binding which causes activation of the second messenger system, adenylyl cyclase cascade, resulting in increased production of cAMP and the initiation of a sequence of membrane and intracellular events. The resulting gene activation leads to increase in testosterone production and male sexual development. Because the same receptor also binds human Chorionic Gonadotropin (hCG), therefore it is called the Luteinising Hormone / Chorionic Gonadotropin Receptor (LH/hCGR), or in short hLHR. The majority of human disorders caused by abnormalities of LH signaling are the results of mutations of the hLHR.
Gain-of-Function Activating Mutations of the LHR – Familial Male-limited Precocious Puberty (FMPP)
In humans, testosterone is almost exclusively (more than 95%) of testicular origin. Under normal physiological conditions, Leydig cells in the testis, are the major source of testicular androgens. The action of LH is mediated by the transmembrane hLHR on the surface of Leydig cells. Because the hLHR when acted on by LH triggers the synthesis of testosterone, therefore we started by examining the hLHR of the FMPP patients. This work led to the identification of single point activating mutations of the hLHR and established the cause of this disease in patients.
FMPP is the result of activating mutations which are gain-of-function mutations and cause luteinising hormone releasing hormone (LHRH) – independent isosexual precocious puberty in boys. Constitutive activity of the hLHR leads to stimulation of testicular Leydig cells in the fetal and prepubertal period in the absence of the hormone resulting in autonomous production of testosterone. At an age as early as 3-4 years old, affected boys have secondary sexual development with penile growth and bilateral enlargement of testes and pubic hair development indistinguishable from true puberty. Traditional diagnosis is made by the findings of pubertal to adult levels of testosterone while the basal level of gonadotropins is appropriate for the age, i.e. prepubertal. The disorder was then called Testotoxicosis because of the early recognition of the production of toxic level of testosterone in prepubertal patients. Histologic examination of testicular biopsy shows growth of Leydig cells2. Activating mutation of the hLHR has no apparent effect on female carriers. To treat this disorder, two drugs are used, spironolactone, an anti-androgen which prevents the action of testosterone and testoleactone, an anti-aromatase which prevents the formation of testosterone. Since upon puberty, there will be a surge of LH secretion which triggers the production of testosterone and sexual development of male, the two drugs are withdrawn upon puberty to allow the naturally produced testosterone to act its course. Thus, Testotoxicosis patients were usually not followed after the onset of natural puberty.
Molecular Diagnosis of FMPP
All known FMPP mutations are single point missense mutations identified in exon 11 of the hLHR gene which encodes the TM domain. The fact that all known activating mutations of hLHR are on a large exon makes molecular diagnosis of FMPP using DNA amplification by PCR followed by sequencing straight forward and relatively simple. Over 120 FMPP kindreds have been reported, more than 50% of which were analysed by my laboratory. Eighteen different mutations of 15 amino acids have been found in patients (Fig 1A)3, 4. One amino acid residue (Asp578) has 3 different mutations while another amino acid residue (Asp564) has 2 different mutations. About 10% of the mutations are sporadic mutations, i.e. new mutations, while there are 10% of FMPP families without identifiable abnormality of the hLHR. The majority of the FMPP mutations were found in TM VI, making this the hot spot of mutation. The most frequently mutated amino acid is Asp-578. Over 55% of all activating mutations affect this amino acid. There is no difference in the clinical manifestation between patients affected by familial and new mutations. Rare mutations occur more often in patients of non-Caucasian ethnic background, including Hispanics, Asians, African Americans and African Brazilians3.
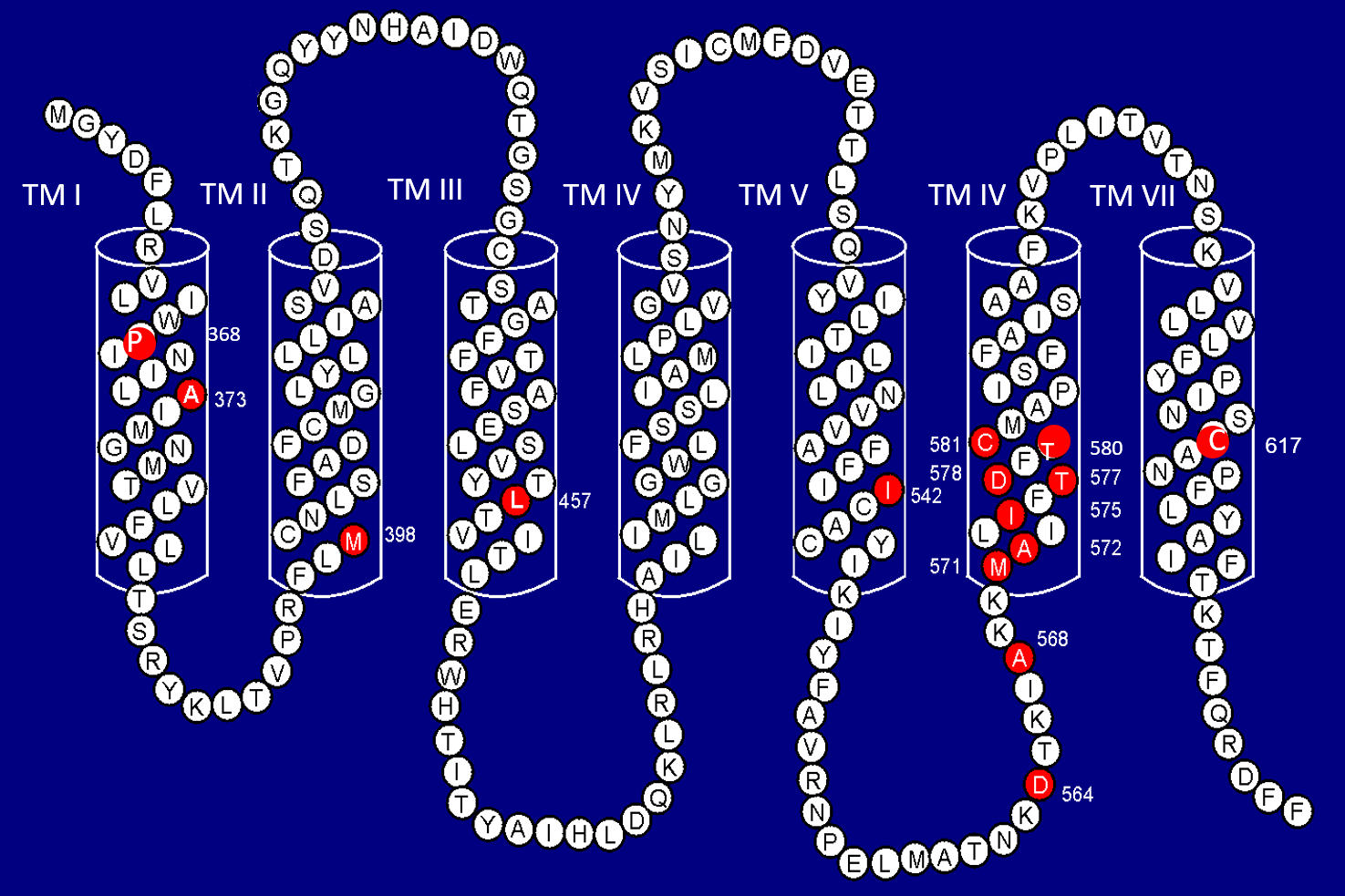
Fig 1A: Human Luteinising Hormone / Chorionic Gonadotropin Receptor (hLHR)
Seven TM Helices (TM I to TM VII). Red filled circles indicates mutated amino acid residues found in FMPP patients. The position of the amino acid residue is indicated by the number.
From Diagnosis to Molecular Mechanism
The mechanism of the activation of the hLHR by the gain-of-function activating mutations is largely unknown. Knowledge from investigating the activating mutation, as well as inactivating mutations of the disease-causing mutations of the hLHR allowed us to conduct molecular modeling study of the hLHR. The majority of the activating mutations are found in TM helices, particularly helix VI of the hLHR. Molecular modeling study and alignment of mutated amino acids on the same side of TM VI suggested that the amino acids affected by the disease-causing mutations of the hLHR are in the interior of the helix bundle pointing towards the inside of the pocket formed by the seven TM helices (Fig 1B and Table 1). Studies with natural mutations of Asp-578 in FMPP patients and site-directed mutagenesis showed that the degree of activation of the hLHR by mutation of Asp-578 is largely determined by the size of the side-chain of the substituting amino acid and not by its charge5. Thus mutation of these amino acids would alter the interhelical interaction and the conformation of the receptor leading to activation. This alteration may simply involve the opening of the pocket formed by the helices. This led us to conclude that Asp-578 is important in serving as a properly positioned hydrogen bond acceptor in the signal transduction activity of the hLHR. Site-directed mutagenesis studies of amino acid residues lying outside of the helices showed that other types of interaction, such as electrostatic and steric, may also be important in receptor activation5.
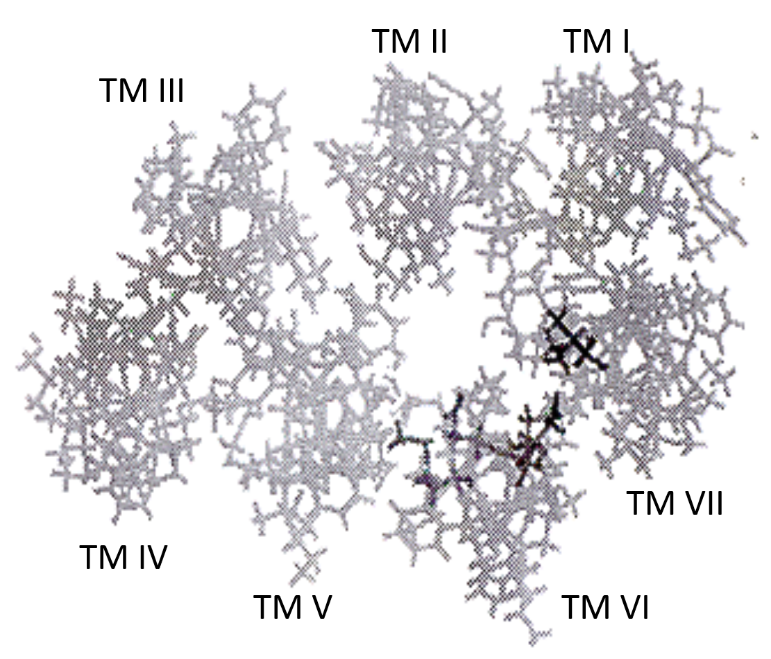
Fig 1B: Human Luteinising Hormone/Chorionic Gonadotropin Receptor (hLHR)
Aerial view of computer model of the TM helices. The darker symbols indicate the mutated amino acid residues.
| Normal AA | Mutation | Position # | Helix Turn # (3.6 AA/Turn) |
Disease |
|---|---|---|---|---|
| Met-571 | Ile | 1 | 0 | FMPP |
| Ala-572 | Val | 2 | 0 | FMPP |
| Ile -575 | Leu | 5 | 1 | FMPP |
| Thr-577 | Ile | 7 | 2 | FMPP |
| Asp-578 | Gly/Tyr/Glu | 8 | 3 | FMPP |
| Cys-581 | Arg | 11 | 4 | FMPP |
| Ile-585 | No known | 14 | 5 | - |
| Ala-589 | No known | 19 | 6 | - |
| Ala-593 | Pro | 23 | 7 | LCH |
Table 1: Alignment of mutated amino acid residues (AA) in TM VI.
No mutation of amino acid residues Ile-585 and Ala-589 has been detected.
The Other Side of the Coin - Loss-of-Function Inactivating Mutations – Leydig Cell Hypolasia (LCH)
Similar missense mutations can also inactivate the receptor and cause subnormal male sexual development. Inactivating mutations are loss-of-function mutations and result in the failure of the receptor to bind the hormone or failure of the bound hormone to activate the receptor. These mutations affect sexual development, especially in the male. In males with homozygous or compound heterozygous inactivating mutations, diminished or abolition of the activity of the hLHR causes resistance to LH stimulation resulting in the failure of testicular Leydig cell differentiation. This gives rise to the anti-thesis of FMPP, Leydig Cell Hypoplasia (LCH) or Leydig Cell Agenesis, a form of pseudohermaphroditism. Clinical presentation of LCH is variable. At one end of the spectrum are genetic male patients with normal-appearing female external genitalia often leading to female sex assignment. At the other end of the spectrum are patients with hypergonadotropic hypogonadism with microphallus and hypoplastic male external genitalia. In between are patients with variable degree of masculinisation of the external genitalia. Since any mutation has the potential of inactivating the protein encoded, the molecular genetics of LCH is more variable than FMPP. The inactivating mutations of the hLHR identified in LCH patients include missense mutations, nonsense mutations, insertions, and deletions. There are also a number of LCH kindreds in which mutations of the hLHR have not been identified indicating that inactivating mutations are very heterogeneous or that LCH is caused by mutation of the hLHR as well as other gene(s). Similar to the variable genetic mutations observed, the effect of the different mutations on the activity of the hLHR is also variable. Depending on the location of the mutation in the receptor protein, it can cause diminished hormone binding, reduced surface expression, abnormal intracellular trafficking, or reduced coupling efficiency, all of which result in reduction or abolition of signal transduction triggered by the binding of the hormone6.
A New Twist - Germ-line and Somatic Activating Mutations in Cancer
Normally, molecular genetic studies of a disorder can be considered to be completed with the design of a diagnostic method and working out of the disease mechanism. However, the studies of FMPP took another turn when an FMPP patient initially diagnosed at 27 months of age at Georgetown University Medical Center basing on endocrine evaluation was found to develop testicular seminoma at the age of 35 years. The diagnosis of FMPP of this patient was confirmed by molecular analysis of the hLHR gene and was found to have the activating mutation Asp578Gly. This was the first report of testicular tumor in a confirmed FMPP patient7. Subsequent to this report, another FMPP patient with the activating mutation Asp564Gly developing two asymptomatic Leydig cell nodules was identified8. The mutations of the hLHR in both cases were germline since the mutations were detected in circulating blood cell genomic DNA. A year after the report of the first FMPP patient developing testicular seminoma Liu et al.9 reported the presence of an activating mutation Asp578His in the hLHR gene in Leydig cell adenoma tissue of three patients. This mutation was not present in adjacent normal tissue or blood cells. Thus, even though high level of testosterone production is not known to cause neoplasia, the presence of an hLHR with constitutive activity might predispose an individual to the development of testicular neoplasia. This discovery changed the traditional practice of releasing FMPP patients after the onset of natural puberty and suggested the necessity of following these patients after puberty in order to track the occurrence of testicular tumor at its early stage.
Mutation-Biased Signaling Pathway of hLHR
Cell Model to Study the Effects of Mutation of hLHR
Asp578His mutation has never been detected in FMPP patients studied and the mutation is somatic and not passed to the patient through the germ cells. The oncogenic mechanism of Asp578His was unknown. These observations opened a new direction in the investigation of the biological effects of activating mutations of the hLHR.
In order to understand the genetic effects of the activating mutations, a cell model was established. A mouse Leydig cell line, MA-10 cells which maintain part of the steroidogenic properties of Leydig cells and respond to LH stimulation was used as a model to compare the effects of the Asp578Gly and Asp578His mutations on Leydig cells. Recombinant pcDNA3 plasmids of wild-type hLHR (hLHR-WT), hLHR carrying the Asp578Gly (hLHR-D578G) or Asp578H (hLHR-D578H) were used to transfect MA-10 cells. Expression profiles of the three types of cells, i.e. MA-10W (MA-10 cells expressing hLHR-WT), MA-10G (MA-10 cells expressing hLHR-D578G) and MA-10H (MA-10 cells expressing hLHR-D578H) were examined using mouse whole genome chips. Genes expressed in the wild-type and the mutant transfectants (3 clones of each category) were analysed by unguided hierarchical clustering as shown in Fig 2A. It is clear clustering separated the expression profile into three groups concordant with the genotype of the expressed hLHR. MA-10G and MA-10H were distinctly different from the control MA-10W cells. There were 132 genes up-regulated in MA-10G and -10H when compared with MA-10W. Most of these were oncogenes and genes related with development, particularly neuronal development. Ontology analyses of the genes differentially expressed between MA-10W and the two mutant cells, MA-10G and MA-10H, identified seven biological categories common to both mutants (Table 2). However, the MA-10G cells revealed two categories of genes, namely those involved in developmental maturation and reproduction, were absent from the MA-10H mutant. This corroborates with the somatic nature of the Asp578His mutation which has not been observed in FMPP patients.
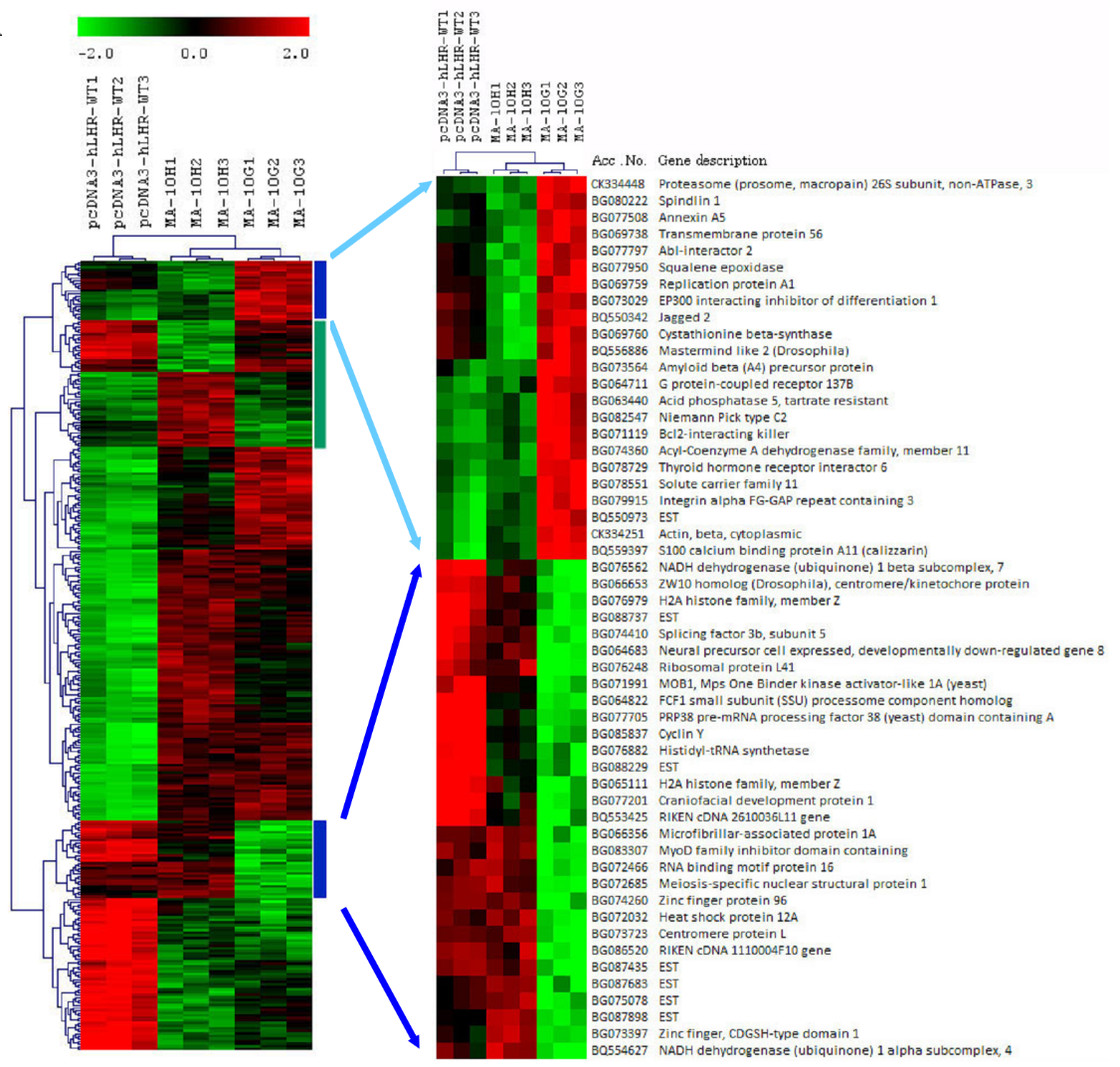
Fig 2A: Hierarchical clustering of MA-10 transfectants and identification of mutation-specific networks.
Hierarchical clustering of expressed genes in MA-10W (pcDNA3-hLHR-WT, clones 1 to 3), MA-10H (hLHR-D578H, clones 1 to 3), and MA-10G (hLHR-D578G, clones 1-3). Blue vertical bar indicates gene clusters with expression signature associated with MA-10G cells ad green vertical bar indicates gene clusters with expression signature associated with MA-10H cells. The blue bar clusters were expanded to show the name of the genes.
| hLHR-Asp578Gly (MA-10G) | hLHR-Asp578His (MA-10H) |
|---|---|
| Cellular process | Cellular process |
| Development | Development |
| Growth | Growth |
| Interaction between organisms | Interaction between organisms |
| Physiological process | Physiological process |
| Regulation of biological process | Regulation of biological process |
| Response to stimulus | Response to stimulus |
| Developmental maturation | - |
| Reproduction | - |
Table 2: Major biological categories based on Gene Ontology analyses expression profiles of MA-10G and MA-10H cells
Comparison of the expression profiles of MA-10G and MA-10H cells revealed two clusters of signature genes differentially expressed between the two mutant cells (Fig 2A, blue bar and green bar), suggesting the genomic effects of the two mutant hLHRs were different. This shows that replacement of the same amino acid by two different amino acids has different effects on the expression of related genes, the first indication of the existence of mutation-biased gene expression with hLHR.
We sought to identify pathways associated with MA-10G and MA-10H mutants by re-generating the cellular networks represented by the gene clusters. To visualise gene expression information in the context of biological networks, the gene clusters were analysed with the GeneGo MetaCore databases integrated from multiple sources, which allowed query and retrieve biologically interesting relations between genes and proteins, and obtained topological properties on integrated networks for biological hypothesis generation. The analysis generated 9 and 12 networks associated with MA-10G cells and MA-10H cells, respectively. To reduce the possibility of convergent networks shared by both mutants, any gene elements common to the two networks were eliminated. This process resulted in two and three networks specific for MA-10G cells and MA-10H cells, respectively (Fig 2B) (Table 3). The mutation-specific networks only involved up-regulated genes.
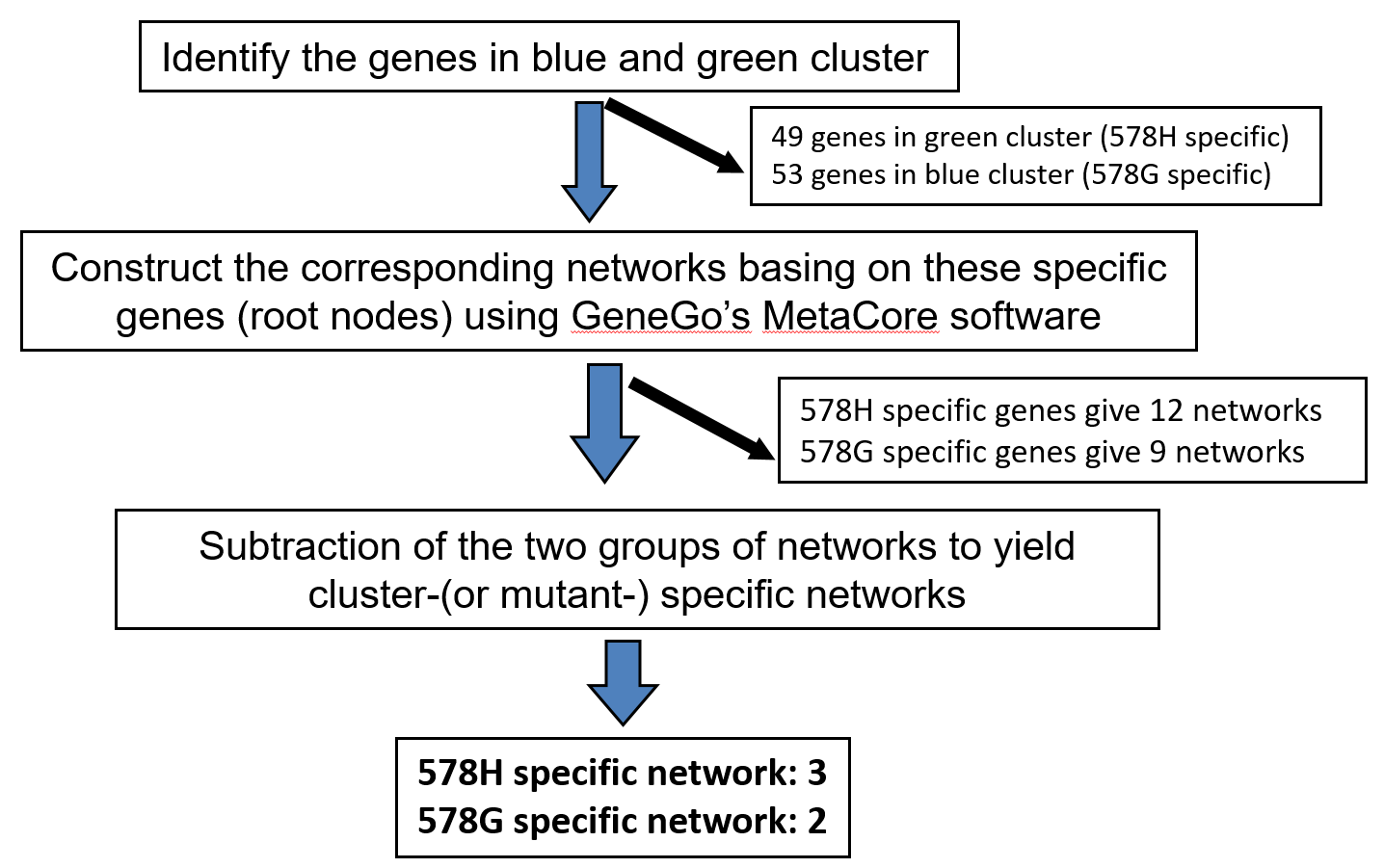
Fig 2B: Hierarchical clustering of MA-10 transfectants and identification of mutation-specific networks.
GeneGo’s MetaCore analysis to identify mutation-specific networks.
| Mutant Genotype | Network | (Regulator) Root Nodes |
|---|---|---|
| Asp578Gly | 1 | (c-Myc) APP、Jagged2、CFDP1、H2AFZ、CBS |
| 2 | (PKC-α) APP-C59, Annexin V | |
| Asp578His | 1 | (c-Src)GUCY1A3,TRIF(TICAM1),Survivin |
| 2 | (nAChR) AARS, Guanylate cyclase, Calcyclin | |
| 3 | (ESR1) MIF, Translin, C8orf40, Adrenomedullin |
Table 3: Mutation-specific networks identified.
Confirmation of Germline hLHR-Asp578Gly-Specific Signaling Network
To confirm the existence of the networks in MA-10G or MA-10H cells, effects of silencing of the key member (regulator) on the other root nodes of the network and on proliferation of the corresponding mutant cells were examined using siRNA. Taking as an example, our results suggested in one of the two MA-10G-specific network, c-Myc, a transcription factor that plays a central role in the control of cellular proliferation, differentiation, apoptosis and cellular maintenance, regulates the expression of down-stream genes with diverse functions related to development such as amyloid beta (A4) precursor protein (App), craniofacial development protein 1 (Cfdp1) and cystathionine beta-synthase (Cbs), and maintenance of DNA structural integrity or gene activation by H2A histone family (H2afz)10 (Fig 3A). MA-10G demonstrated higher basal c-Myc activity compared to MA-10W (56%) and MA-10H cells (64%), and it was more sensitive to c-Myc siRNA inhibition by revealing 40% decrease in activity (p < 0.05) (Fig 3B). C-Myc was regulated by Notch1 which was a downstream target of Jagged2, a gene up-regulated by hLHR-D578G as revealed by expression profiling. The regulation of Notch1 by Jagged2 was demonstrated by the Western blotting results showing the suppression of Notch1 represented by decreased cleaved Notch1 level in MA-10G cells after treatment with Jagged2 siRNA but not in MA-10H cells which did not have much Notch1 protein to start with (Fig 3C). The activity of the c-Myc network in MA-10G cells was confirmed by the experiment which showed that silencing of c-Myc caused suppression of the expression of all components of the c-Myc network, namely, App, Cfdp1, H2afz, and Cbs (Fig 3D). Fig 3E further showed that proliferation of MA-10G cells was more sensitive to c-Myc siRNA than c-Src siRNA (control), indicating the relative importance of the c-Myc pathway in these cells. Collectively, these experiments demonstrated that one of the hLHR-D578G specific pathways was the up-regulation of Jagged2 by germline activating mutation, which served as the switch for Notch1 – c-Myc regulation and led to increased cell proliferation.
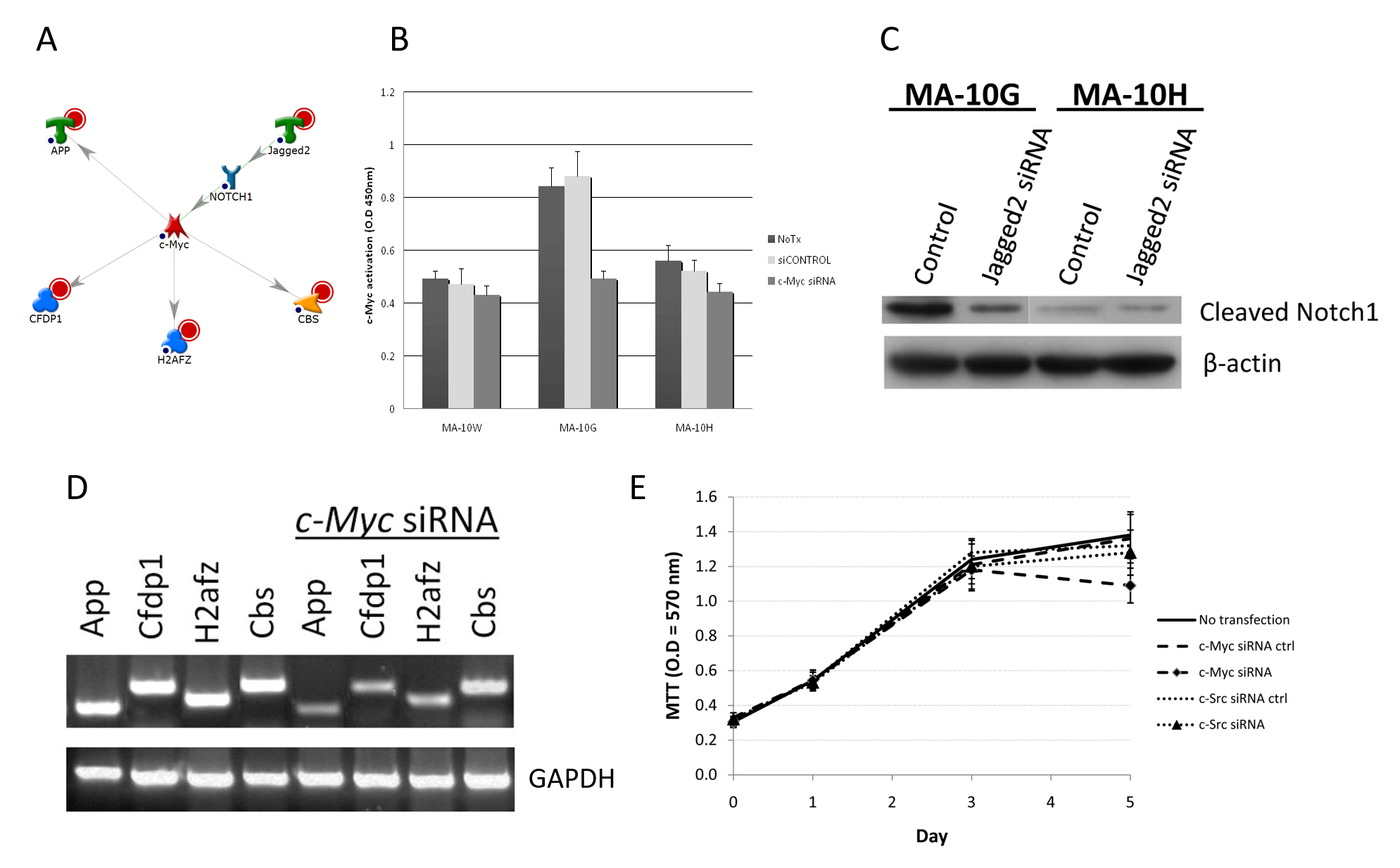
Fig 3A-E: Confirmation of Germline hLHR-Asp578Gly-Specific c-Myc Signaling Network.
A: c-Myc network in MA-10G cells. Root nodes were indicated by red circles. B: Effect of c-Myc siRNA on c-Myc activity in MA-10G cells. Inhibition of c-Myc and c-Src was performed by transient transfection of 100nM of ON-TARGETplus siRNA (Dharmacon, Chicago, IL) for 24 hrs based on manufacture’s protocol. To confirm silencing specificity of individual siRNAs, ON-TARGETplus siCONTROL non-targeting control (Dharmacon, Chicago, IL) was also included in each transfection experiment. Activated c-Myc and c-Src signaling proteins were quantified using TransAM c-Myc and FACE c-Src kit (Active Motif, Carlsbad, CA). The activation signal was measured by colourimetric reaction with optical density set at 450 nM using spectrophotometer. MA-10G demonstrated higher basal c-Myc activity and more sensitive to c-Myc inhibition by revealing 40% decrease in activity (p < 0.05). C: Confirmation of regulation of c-Myc by Jagged2 mediated by Notch1. Notch1 involvement in MA-10G cells implicated in GO and network analysis was examined for regulation by Jagged2. Upon ligand binding Notch receptors are cleaved and release an intracellular subunit that migrates to the nucleus where it acts as a transcriptional coactivator. Therefore the amount of cleaved Notch1 was examined with or without Jagged2 inhibition by Western blot in both MA-10G and MA-10H cells. Noticeable decrease on cleaved Notch1 was observed in MA-10G but not MA-10H cells. D: Regulations of downstream genes by c-Myc. Cells were transfected with c-Myc siRNA followed by semi-quantitative RT-PCR. Expression of App, Cfdp1, H2afz and Cbs was examined before and after siRNA transfections. Silencing of c-Myc caused suppression of App, Cfdp1, H2afz, and Cbs expression indicating the operation of all components of the c-Myc network in MA-10G. E: To study the specific effect of c-Myc on cell proliferation with either wild or mutated hLHR, a 5-day 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was conducted with c-Myc siRNA transfection. Proliferation of MA-10G was more sensitive to c-Myc siRNA than c-Src siRNA (control), indicating the relative importance of the c-Myc pathway in MA-10G cells.
Confirmation of Somatic hLHR-Asp578His-Specific Signaling Network
In one of the three MA-10H-specific networks, H-specific cluster genes guanylate cyclase 1 alpha 3 (Gucy1a3), toll-like receptor adaptor molecule (Ticam1) and Survivin (Birc5) were regulated through the control of c-Src, a known G-protein receptor target and regulator of transcription factor NF-κB (Fig 4A). C-Src (proto-oncogene tyrosine-protein kinase Src) is a lipid-modified signaling molecule (myristoylated) involved in cell-cell interactions, cell migration and proliferation through the phosphorylation of many substrates11. In the case of MA-10H cells, higher c-Src activity was found compared to MA-10W and MA-10G by 26% and 27%, respectively (Fig 4B). Even though transfection with c-Src siRNA led to decrease in c-Src activity in MA-10G cells, MA-10H cells were more sensitive to the same treatment by showing 33% decrease in activity (P < 0.05). This finding together with the previous study on c-Myc confirmed the predominant regulatory role of c-Myc in MA-10G and c-Src in MA-10H cells, respectively (Fig 4B). Proliferation of MA-10H cells was more sensitive to c-Src siRNA than c-Myc siRNA (control), indicating the relative importance of the c-Src pathway in these cells (Fig 4C). Functioning of the c-Src pathway in MA-10H cells was further corroborated by the demonstration that silencing of c-Src, but not c-Myc, caused suppression of Gucy1a3, Ticam1, and Birc5 (Survivin) expression – all components of c-Src network (Fig 4D). Collectively, these experiments confirmed that the c-Src pathway played a prominent role in signal transduction of the LH with the Asp578His mutation.
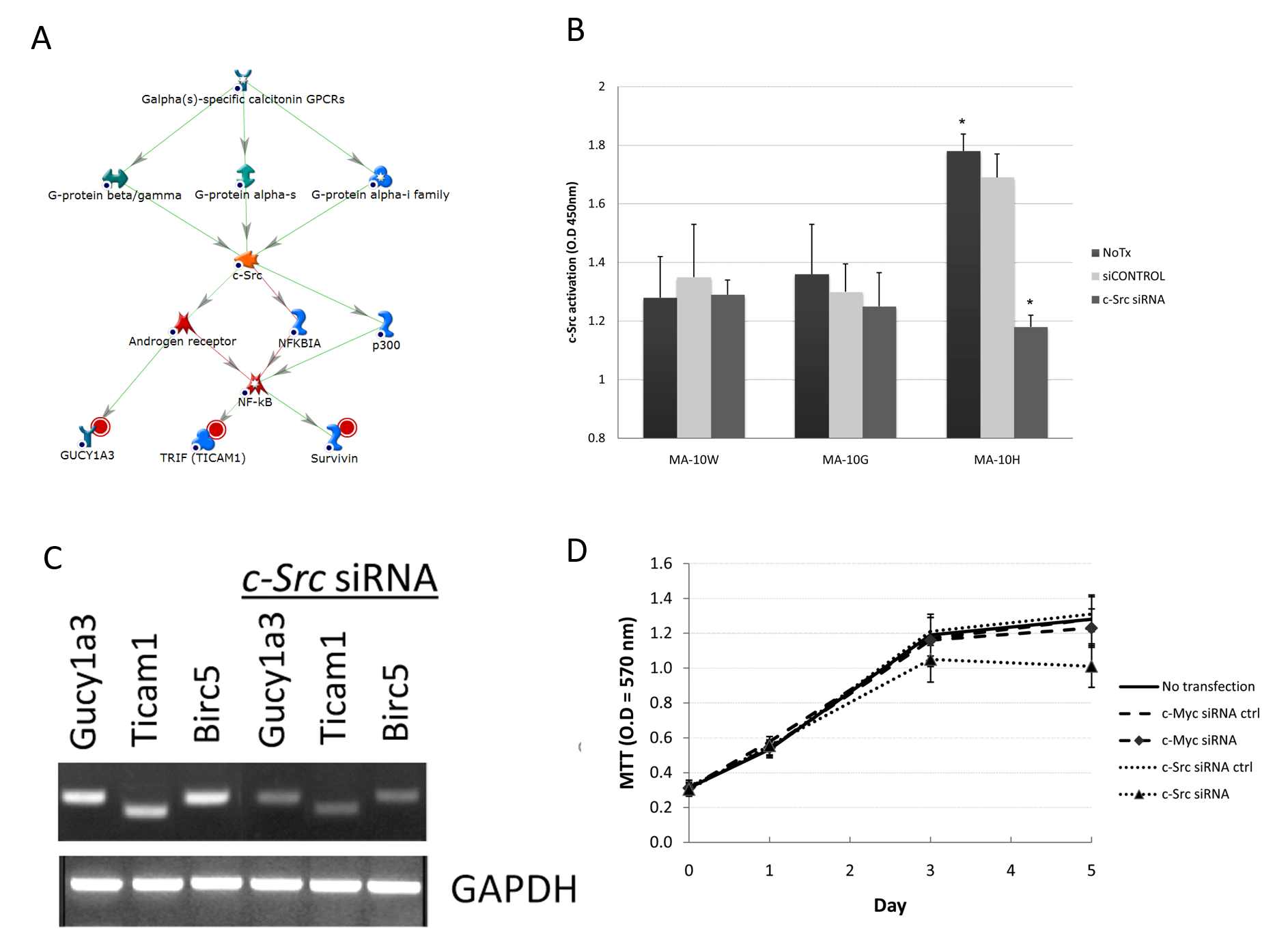
Fig 4A-D: Confirmation of Somatic hLHR-Asp578His-Specific c-Src Signaling Network.
A: c-Src network in MA-10H cells. Root nodes were indicated by red circles. B: Effect of c-Myc siRNA on c-Myc activity in MA-10G cells. Inhibition of c-Myc and c-Src was performed by transient transfection of 100nM of ON-TARGETplus siRNA (Dharmacon, Chicago, IL) as described under Fig 3B. Higher c-Src activity was found compared to MA-10W and MA-10G. Even though transfection with c-Src siRNA led to decrease in c-Src activity in MA-10G cells, MA-10H cells were more sensitive to the same treatment by showing more than 33% decrease in activity (P < 0.05). C: Regulations of downstream genes by c-Src. Cells were transfected with c-Src siRNA followed by semi-quantitative RT-PCR. Expression of Gucy1a3, Ticam1, and Birc5 (Survivin) was examined before and after siRNA transfections. Silencing of c-Src caused suppression of Gucy1a3, Ticam1, and Birc5 (Survivin) expression indicating the operation of all components of c-Src network in MA-10H. D: To study the specific effect of c-Src on cell proliferation as described under Fig 3E. Results showed that proliferation of MA-10H was more sensitive to c-Src siRNA than c-Myc siRNA (control), indicating the relative importance of the c-Src pathway in MA-10H cells.
Conclusion
The story started with the goal of delineating the etiology of FMPP, a disorder caused by abnormal production of testosterone leading to precocious puberty in boys. Knowledge of the physiology of testosterone production directed our attention to the disconnection between hLH level and testosterone biosynthesis and the discovery of the activating mutations of the hLHR, the mediator of hLH and the testosterone synthesis machinery. Subsequently, this led to the establishment of a molecular diagnostic method that allows accurate diagnosis for over 90% of all cases of FMPP. Extensive studies of the molecular genetics of FMPP patients and computer modeling of the hLHR led to the tentative definition of the mechanism of signal transduction by the transmembrane helices of the hLHR. The occurrence of testicular tumor in a returning patient whose FMPP status was confirmed with the newly established molecular diagnostic method ignited a new search for the potential connection between FMPP, hLHR mutation, and testicular tumor. This subsequently led to the change in the management of FMPP patients and suggested the necessity of frequent recall of patients after puberty for the early detection of testicular tumor. This also led to the discovery of two classes of activating mutations of the hLHR, namely, germline mutations and somatic mutation. The somatic mutation of hLHR, Asp578His does not lead to FMPP, but instead the development of testicular tumor. More recent expression profiling and cell model studies revealed the presence of mutation-specific pathways of an amino acid (578Asp) which could explain the germline and somatic nature of the specific mutations. These twists and turns in the course of study of the hLHR in FMPP were not anticipated in the beginning. The excitement accompanying the unfolding of the story is perhaps part of the rewards that drive scientific discovery.
Acknowledgments
This work is supported in part by the Hong Kong Branch of CAS Center for Excellence in Animal Evolution and Genetics, VCDF CUHK No. 8601011
References
- Dufau ML. The luteinising hormone receptor. In: The Leydig Cell (Payne AH, Hardy MP, Russell LD, Eds). Cache River Press, IL, pp. 333-350, 1996.
- Holland FJ. Gonadotropin-independent precocious puberty. Endocrinol Metab Clin North Amer. 20:191–210, 1991.
- Wu SM, Leschek EW, Rennert OM, Chan WY. Luteinising hormone receptor mutations in disorders of sexual development and cancer. Frontiers Biosci. 5:D342-352, 2000.
- Nagasaki K, Katsumata N, Ogawa Y, Kikuchi T, Uchiyama M: Novel C617Y mutation in the 7th transmembrane segment of luteinising hormone/choriogonadotropin receptor in a Japanese boy with peripheral precocious puberty. Endocr J. 57(12):1055-60, 2010.
- Wu SM, Leschek EW, Brain C, Chan WY. A novel activating mutation of the luteinising hormone/chorionic gonadotropin receptor in a patient with FMPP - Effects of size versus charge. Mol Genet Metab. 66:68-73, 1999.
- Wu SM, Chan WY. Male pseudohermaphroditism due to inactivating luteinising hormone receptor mutations. Arch Med Res. 30:495-500, 1999.
- Martin MM, Wu SM, Martin ALA, Rennert OM, Chan WY. Testicular seminoma in a patient with a constitutively activating mutation of the luteinising hormone/chorionic gonadotropin receptor. Euro J Endocrinol. 139:101-106, 1998.
- Leschek EW, Chan WY, Diamond D, Laefer M, Jones J, Barnes KM, Cutler GB Jr. Nodular leydig cell hyperplasia in a boy with familial male-limited precocious puberty (FMPP). J Pediatr. 138:949-951, 2001.
- Liu G, Duranteau L, Monroe J, Doyle DA, Carel JC, Shenker A. Leydig-cell tumors caused by an activating mutation of the gene encoding the luteinising hormone receptor. N Engl J Med. 341:1731-1735, 1999.
- Dang CV. Myc, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. Aug; 3(8): a014217, 2013.
- Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell. 6(3):209-214, 2004.
Author: Prof Chan Wai-yee
BSc (CUHK), PhD (FLA)
Pro-Vice-Chancellor / Vice-President, CUHK
Master, CW Chu College, CUHK
Li Ka Shing Professor of Biomedical Sciences, CUHK
Prof Chan obtained his B.Sc. (Hon. 1st Class) in Chemistry from The Chinese University of Hong Kong in 1974 and Ph.D. in Biochemistry from the University of Florida, Gainesville, Florida, USA in 1977. He served as tenured Assistant to Associate Professor at the Department of Pediatrics and Department of Biochemistry and Molecular Biology, University of Oklahoma Health Science Center, Oklahoma City, OK, USA from 1979-1989, and tenured Professor, Department of Pediatrics and Department of Biochemistry, Cell & Molecular Biology, Georgetown University Medical Center, Washington, DC, USA from 1989 to 2009. He was jointly appointed at the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institutes of Health (NIH), USA in 2001 and helped found the Laboratory of Clinical Genomics. He was later appointed as Head and Principal Investigator of the Section on Developmental Genomics, NICHD. In June 2009 he became Chair Professor of Biomedical Sciences and Director of the School of Biomedical Sciences, Faculty of Medicine, The Chinese University of Hong Kong. Currently he is also the Director of the Institute for Tissue Engineering and Regenerative Medicine, CUHK, and Director of CUHK-BGI (Beijing Genome Institute) Innovation Institute of Trans-Omics. He also serves as Director for several joint laboratories including the CUHK-Shandong University Joint Laboratory on Reproductive Genetics, CUHK-Chinese Academy of Science (CAS) Guangzhou Institute of Biomedicine and Health Joint Research Laboratory on Stem Cell and Regeneration, CUHK-CAS Kunming Institute of Zoology Joint Laboratory of Bioresources and Molecular Research in Common Disease, and Ministry of Education (MOE) Key Laboratory (CUHK-Jinan University) on Regenerative Medicine. He has been serving as Master of CW Chu College since January, 2017.
Prof Chan's expertise is in the molecular genetics of human reproductive and endocrine disorders, biochemical genetics of inborn errors of copper and zinc metabolism, functional genomics and epigenomics of germinal stem cells and reproductive tumors, and non-coding RNAs in the differentiation of human induced pluripotent stem cells (iPSCs). His research resulted in 7 patents, 5 books, 29 book chapters, and over 200 articles published in international peer-reviewed scientific journals.
Prof Chan serves as editor / associate editor of 8 international scientific journals and as member of the editorial board of 16 others. He is a reviewer for a number of research granting agencies including the National Institutes of Health, USA, the Wellcome Trust and Health Research Board, UK, the Science Foundation of Ireland, the French National Alliance for Life and Health Sciences and French National Cancer Institute, INSERM, France, and the Macau Science and Technology Development Fund, Macau SAR. He is also a member of the Grant Review Board, Food and Health Bureau and the Biology and Medicine Panel of the Research Grants Council, University Grants Committee, Hong Kong SAR. Prof Chan is active in the community, serving as member in several public offices, including as a Specialist for the Hong Kong Council for Accreditation of Academic and Vocational Qualifications. He has also served in a number of professional academic societies, including as President of the Association of Chinese Geneticists in America, USA and as a member of the Development Committee of the Society for the Study of Reproduction, USA. Currently he is the Executive Director of the Association of Chinese Geneticists in America, USA, a member of the Board of Directors of Joshua Hellmann Foundation for Orphan Diseases, Hong Kong SAR, and a member of the Council of the Shaw Prize Foundation.
Prof Chan has received a number of honours including Honorary Member of the Chinese American Medical Association, New York, in 1992, a Distinguished Faculty Tutor, School of Medicine, Georgetown University in 1997 and a Distinguished Mentor Nominee, National Institutes of Health, in 2005 and 2006. He was also a recipient of the 1988 Merrick Award for Outstanding Biomedical Research from the Oklahoma Medical Research Foundation, USA, 1989 Service Award from the University of Oklahoma Health Sciences Center, USA, and the 2008 Presidential Award from the Association of Chinese Geneticists in America, USA.
November 2020